
Abstract
Prion diseases comprise a group of rapidly progressive and invariably fatal neurodegenerative disorders for which there are no effective treatments. While conversion of the cellular prion protein (PrPC) to a β-sheet rich isoform (PrPSc) is known to be a critical event in propagation of infectious prions, the identity of the neurotoxic form of PrP and its mechanism of action remain unclear. Insights into this mechanism have been provided by studying PrP molecules harboring deletions and point mutations in the conserved central region, encompassing residues 105–125. When expressed in transgenic mice, PrP deleted for these residues (Δ105–125) causes a spontaneous neurodegenerative illness that is reversed by co-expression of wild-type PrP. In cultured cells, Δ105–125 PrP confers hypersensitivity to certain cationic antibiotics and induces spontaneous ion channel activity that can be recorded by electrophysiological techniques. We have utilized these drug-hypersensitization and current-inducing activities to identify which PrP domains and subcellular locations are required for toxicity. We present an ion channel model for the toxicity of Δ105–125 PrP and related mutants and speculate how a similar mechanism could mediate PrPSc-associated toxicity. Therapeutic regimens designed to inhibit prion-induced toxicity, as well as formation of PrPSc, may prove to be the most clinically beneficial.
Acknowledgments
Work in the Harris laboratory is supported by National Institutes of Health Grants NS052526 and NS040975 (to D.A.H.). I.H.S. is supported by National Institutes of Health Pre-doctoral Fellowship NS063547, and by the Medical Scientist Training Program at Washington University.
Figures and Tables
Figure 1 Spontaneous inward currents induced by expression of ΔCR PrP in HEK cells. HEK cells expressing vector, WT PrP, ΔCR PrP or ΔCR + WT PrP were analyzed by whole-cell patch-clamping. Recordings were made at a holding potential of −80 mV. Spontaneous current activity was observed only in cells expressing ΔCR PrP, and was suppressed by co-expression of WT PrP.
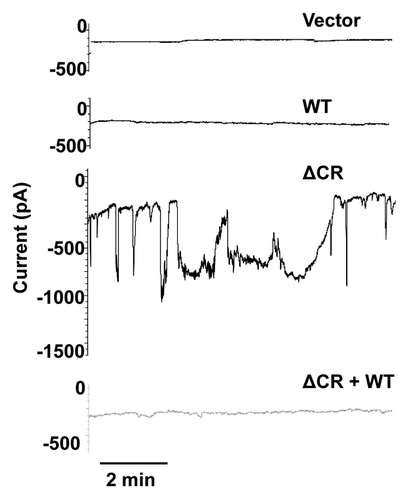
Figure 2 Schematic of murine PrP showing domain structure and mutations. ER signal sequence (SS, blue, aa 1–22); polybasic domain (yellow, aa 23–31); octapeptide repeats (OR, magenta, aa 51–90); charge cluster (CC, red, aa 94–110); hydrophobic domain (HD, green, aa 111–134); GPI signal sequence (GPI, purple, 231–254). Three α helices (H1, H2 and H3), two N-linked oligosaccharides (hexagons), and one disulfide bond (S-S) are present in the C-terminal half of PrP. The sequences for amino acids 23–31 and 94–134 are shown above the schematic. The boxed residues (105–125) indicate those deleted in ΔCR PrP. Residues mutated in familial prion diseases are indicated in red, and a nonpathogenic polymorphism in blue, with arrows pointing to the substituted amino acid.
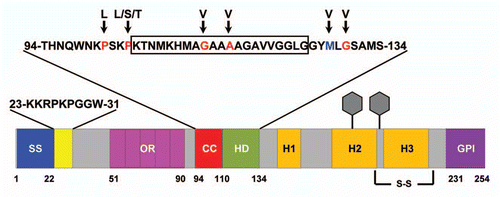
Figure 3 Ion Channel Formation Model of PrP-dependent toxicity. (A) ΔCR PrP, as a result of deletion of amino acids 105–125 (red), creates channels or pores in the plasma membrane by insertion of N-terminal, polybasic region (residues 23–31, shown in yellow). This allows cations (purple balls) to permeate the plasma membrane. The presence of WT PrP closes the channel. (B) PrPSc (orange) or oligomers of the Aβ peptide (red) bind to PrPC and induce a ΔCR-like conformational change, resulting in transient pore formation and toxicity.
Table 1 Summary of structure-function study