Abstract
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease caused by a selective loss of motor neurons. There is no cure and few effective treatments. The RNA-binding protein TDP-43 contributes to the pathogenesis of ALS. TDP-43 is depleted from the nucleus and accumulates in cytoplasmic aggregates in the degenerating neurons and glia of most ALS patients. Furthermore, mutations in the TDP-43 gene cause rare familial and sporadic forms of the disease. Thus, therapeutic strategies targeting TDP-43 may be efficacious. We have used the yeast model system to identify the mechanisms by which TDP-43 aggregation contributes to ALS and to identify approaches to protect cells from the toxic effects of TDP-43 aggregation. Using an unbiased yeast genetic screen we discovered Dbr1 as a potent suppressor of TDP-43 toxicity. Yeast cells in which Dbr1 is deleted are resistant to TDP-43 toxicity. Dbr1 inhibition in mammalian cells is also sufficient to protect against TDP-43 cytotoxicity. Here, we review this recent discovery, highlighting future approaches aimed at extending these studies and pursuing Dbr1 as a novel therapeutic target for ALS.
Keywords: :
Amyotrophic lateral sclerosis (ALS), commonly known as Lou Gehrig's disease, is a devastating neurodegenerative disease that selectively affects cortical, brainstem and spinal motor neurons. Motor neuron degeneration occurs with rapid progression, resulting in muscle paralysis and death typically within 3–5 y of disease onset. The causes of about 90% of ALS cases remain unknown. However, remarkable genetic advances have provided a foot in the door into the pathogenesis of ALS. As of 2013, over 15 ALS disease genes have been identified.Citation1 Approximately 5–10% of cases show a Mendelian pattern of inheritance and occur within multigenerational affected families (familial ALS, or fALS). However, the vast majority of patients lack a clear family disease history and exhibit a sporadic disease occurrence (sporadic ALS, or sALS).
In 1993, mutations in the SOD1 gene encoding superoxide dismutase 1 were identified as the first known cause of ALS.Citation2 A gain-of-function hypothesis for mutant SOD1 is now widely accepted. While there have been many reports of successful pharmacologic and genetic modifiers of motor neuron loss and disease progression in transgenic mice carrying the mutant human SOD1 gene, these targets have largely failed to mitigate the disease when translated into human clinical trials.Citation3 There are currently no effective therapies for stopping the disease, and only one drug, Riluzole, has been approved to modify disease progression merely by a few months.Citation4 Thus, novel therapeutic strategies are desperately needed. The elucidation of additional genetic contributors to ALS will illuminate new disease pathways and help to suggest new directions and targets for the development of therapies.
After several years of intense focus on SOD1, the ALS research field was dramatically re-focused by a major breakthrough in 2006, when the DNA- and RNA-binding protein TDP-43 (TAR DNA-binding protein of 43 kDa) was identified as the major component of ubiquitylated cytoplasmic inclusions in affected patient spinal cord neurons.Citation5 These inclusions are the hallmark of ALS neuropathology in almost all ALS cases (except those harboring SOD1 mutations) along with a marked clearance of TDP-43 from the nucleus. Soon after it was revealed that several mutations in the TARDBP gene, which encodes TDP-43, can cause some rare forms of fALS and sALS.Citation6,Citation7 TDP-43 inclusions are also prevalent in two other rare, progressive neurodegenerative diseases: Inclusion Body Myopathy with Paget disease of bone and frontotemporal dementia (IBMPFD) and Perry syndrome, as well as in secondary pathology in Huntington, Parkinson and Alzheimer diseases.Citation8 The conjunction of genetics and pathology place TDP-43 as a major player in ALS and related neurodegenerative diseases. Shortly following the discovery of a major role of TDP-43 in ALS, several additional studies unleashed a paradigm shift in ALS research, with intense focus on a role of RNA metabolism and defects in RNA processing pathways. These include the discovery of disease-causing mutations in another RNA-binding protein gene, FUS/TLS, as a cause of fALS,Citation6 an association between intermediate-length polyglutamine expansions in ataxin 2 (which functions in RNA-metabolism) and increased risk for ALS,Citation9 and potential toxic RNA resulting from large GGGGCC repeat expansions in the C9orf72 gene,Citation10,Citation11 which appears to be the most common cause of ALS. Hence, RNA dysregulation is widely hypothesized to be a central component to disease onset and progressionCitation12 and thus opens up many new possibilities about potential avenues to pursue for therapeutic intervention.
Genetic model systems are the experimental workhorses for elucidating key mechanisms and pathways underpinning many aspects of biology with direct relevance to human disease.Citation13 Recent experimental results in several model organisms, including yeast, worms, flies and mice have helped to define key features of TDP-43 that may be important for disease pathogenesis. These model systems also provide a platform for genetic and chemical screens to reveal modifiers. Our laboratory has used the budding yeast Saccharomyces cerevisiae as a genetically-tractable model to study the effects of TDP-43 and FUS aggregation.Citation14,Citation15 Though seemingly simple, the yeast model system offers many powerful experimental advantages, which have been brought to bear on many facets of cell biology, several with direct connections to human disease. Yeast models have revealed unexpected new insights into the mechanisms of other neurodegenerative diseases, including Parkinson disease,Citation16-Citation19 Huntington disease,Citation17,Citation20,Citation21 and Alzheimer diseaseCitation22 and have also provided a platform for the discovery of chemical compounds that modify proteotoxicityCitation23,Citation24
Expressing human WT TDP-43 at low levels in yeast cells results in nuclear localization. Increasing the expression level of TDP-43 (for example, high copy plasmid or stronger promoter) results in the formation of numerous cytoplasmic TDP-43 foci and growth inhibition.Citation14 Thus, this simple model system is able to recapitulate two cardinal features of TDP-43 proteinopathy: cytoplasmic aggregation and cytotoxicity. Since full-length TDP-43 aggregated in the cytoplasm and was toxic, we generated a panel of truncation constructs to define the regions of TDP-43 that were sufficient and necessary for aggregation and toxicity. We found that the C-terminal domain of TDP-43 was absolutely required for aggregation and toxicity, but, interestingly, was not sufficient: an intact RNA-recognition motif (RRM) was also needed. Hence, RNA-binding seemed to be a component of TDP-43 toxicity.Citation14 These findings are significant for two main reasons. First, it implicates the C-terminal domain of TDP-43 as an important driver of TDP-43 aggregation. We went on to confirm a direct role for the C-terminal domain in TDP-43 aggregation using in vitro assays with recombinant TDP-43.Citation25 It is remarkable that of the over 30 ALS-linked TDP-43 mutations that have been reported since 2008, all but one of them are located in the C-terminal domain, underscoring the power of the yeast model system for elucidating important disease-relevant features of TDP-43. Second, the requirement for RNA-binding as a component of TDP-43 toxicity (preventing RNA-binding via truncation or point mutations abolishes TDP-43 toxicity) was interesting but confusing at the time we discovered it. However, it has since been confirmed by others in multiple model systems, including fly, worm and mammalian cells. Moreover, it has provided the framework for recent high impact genomewide approaches aimed at cataloging all of the RNA targets of TDP-43.Citation26
When ALS-linked mutations in TDP-43 were discovered, it was unclear how these mutations contributed to disease. Did they cause a loss of TDP-43 function? Did they make the protein more aggregation-prone? Over 30 pathogenic mutations in the TDP-43 gene have been identified in ALS patients. We tested over 20 of these in the yeast model and in vitro aggregation system. We discovered that TDP-43 is inherently aggregation-prone and ALS-linked mutations accelerate aggregation and enhance toxicity.Citation25 Importantly, several other laboratories have also seen similar effects of ALS-linked mutations on TDP-43 in diverse experimental systems ranging from cell culture, flies, chicken embryos, mouse, zebrafish and rat,Citation27-Citation33 again underscoring the utility of the yeast model system for uncovering disease-relevant insight into TDP-43 pathobiology.
The yeast TDP-43 model and the initial studies described above allowed us to define key features of TDP-43 that were relevant to disease and to better understand what drives TDP-43 aggregation. But the big challenge was to discover the mechanism by which TDP-43 is toxic. We reasoned that we could harness the yeast model system to perform unbiased genomewide modifier screens to elucidate the cellular pathways affected by TDP-43 aggregation and toxicity. In other words, if we could find genes that could suppress or enhance TDP-43 toxicity it might tell us why TDP-43 was toxic in the first place. Accordingly, we have used the TDP-43 yeast model to perform high-throughput screens and identified several potent modifiers of TDP-43 toxicity, some with human homologs.
We have now completed two TDP-43 modifier screens: a yeast plasmid overexpression screen and a yeast deletion screen. One of the hits from the plasmid overexpression screen (overexpress each yeast gene to look for suppressors and enhancers of TDP-43 toxicity) was PBP1, which is the homolog of a human neurodegenerative disease protein, ataxin 2. Overexpression of Pbp1 enhanced TDP-43 toxicity and deletion of Pbp1 suppressed toxicity.Citation9 We validated this genetic interaction in the fly nervous system and showed TDP-43 and ataxin 2 physically associate in an RNA-dependent manner in mammalian cells. We also found that the ataxin 2 protein is mislocalized in ALS patient spinal cord neurons. Ataxin 2 is a polyglutamine (polyQ) disease protein. The polyQ tract of ataxin 2 is normally 22 or 23 Qs and polyQ expansions > 34 cause spinocerebellar ataxia 2 (SCA2). Given the striking interactions between ataxin 2 and TDP-43, we hypothesized that polyQ expansions in ataxin 2, which were longer than normal but not long enough to cause SCA2, might be associated with ALS. To test this hypothesis we analyzed the ataxin 2 gene (ATXN2) in 915 individuals with ALS and 980 healthy controls and found intermediate-length polyQ expansions in ataxin 2 (27–33Q) significantly associated with increased risk for ALS. We find these expansions in ~5% of ALS patients, thus making this one of the most common genetic risk factors for ALS discovered to date.Citation9 Importantly, this association between ataxin 2 intermediate-length polyQ expansions and ALS has been confirmed by numerous other laboratories in diverse ALS patient populations worldwide.Citation34-Citation37 The discovery of one of the most common genetic risk factors for ALS starting from a simple yeast screen highlights the usefulness of this model system and approach for gaining important insight with direct relevance to human disease.
In addition to these genetic screens providing insight into disease mechanisms and new disease genes and risk factors, we have also unexpectedly revealed a novel potential therapeutic target for ALS. In a complementary genetic screen (testing each non-essential yeast gene deletion for enhancement or suppression of TDP-43 toxicity), one of the most effective gene deletion suppressors of TDP-43 toxicity is a yeast gene called DBR1, which encodes RNA lariat debranching enzyme and is conserved from yeast to human.Citation38 Dbr1 functions as a phosphodiesterase, linearizing RNA lariats from their circular form after intronic splicing of pre-mRNA (). In the absence of Dbr1 function, RNA lariats are not debranched, a step necessary for their degradation by exonucleases in the cell. Thus, the lariats accumulate at high levels in the cell.Citation39,Citation40 If yeast or mammalian Dbr1 is restored to the cell, lariats are debranched and TDP-43’s toxicity returns.Citation38 Using a panel of Dbr1 mutants with varying degrees of enzymatic activity, we showed that TDP-43 toxicity correlates with relative accumulation of RNA lariat species.Citation38
Figure 1. RNA lariats decoy cytoplasmic TDP-43 aggregates. (A) When introns are spliced out of pre-mRNAs, Dbr1 debranches the excised lariat introns and then they are degraded by cellular exonucleases. In the absence of Dbr1 activity, intronic lariats accumulate in the cytoplasm. (B) A hypothetical model of how Dbr1 inhibitors could be pursued as a therapeutic strategy for ALS. In a healthy motor neuron, TDP-43 is localized to the nucleus. In ALS, TDP-43 is lost from the nucleus and accumulates in the cytoplasm where it forms large aggregates. These aggregates might bind and inhibit essential cellular RNAs and RNA-binding proteins. Inhibiting Dbr1 would lead to the accumulation of RNA lariats in the cytoplasm and these might act as decoys for TDP-43, relieving the inhibition of the essential RNAs and RNA-binding proteins.
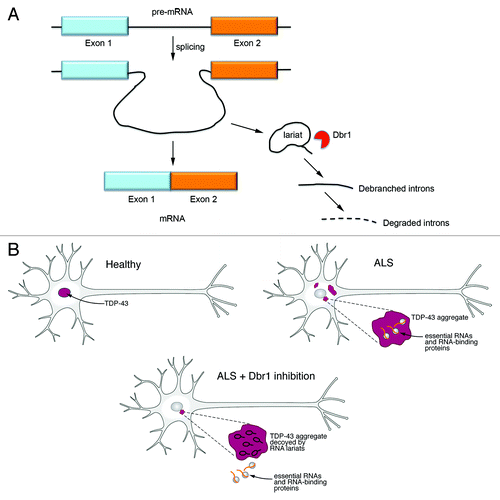
Given TDP-43’s affinity for intronic sequences,Citation26 we hypothesized that these accumulated lariats might act as a kind of decoy and sequester TDP-43 away from interfering with important cellular RNAs and RNA-binding proteins (). To test this hypothesis, we developed a method to visualize intronic lariats in living cells.Citation38 We modified the m-TAG technique, which is used to visualize mRNA localization in diverse cell types.Citation41 Instead of visualizing mRNAs, we were interested in visualizing the localization of intronic lariats. Using homologous recombination, we inserted binding sites for the MS2 viral coat-protein into the intron of the endogenous ACT1 yeast gene, which is known to accumulate as RNA lariats in the absence of Dbr1.Citation40 Expression of MS2 viral coat protein fused to GFP (MS2-CP-GFP), which has high affinity for the MS2 stem-loop sequence, allowed us to visualize the accumulation of intronic lariats only in the absence of Dbr1.Citation38 The lariat RNAs consistently formed one or two bright foci in dbr1Δ yeast cells but remained diffuse and faint in wild-type cells. To test our hypothesis, we expressed TDP-43 in these WT and dbr1∆ yeast cells and used immunocytochemistry to detect TDP-43 and fluorescence microscopy to visualize the GFP-tagged intronic lariats. Remarkably, the TDP-43 cytoplasmic aggregates perfectly co-localized with the fluorescently labeled lariats,Citation38 supporting the hypothesis that TDP-43 cytoplasmic aggregates are preferentially sequestered by intronic lariats that accumulate upon Dbr1 inhibition.
We extended our findings from yeast to mammalian cells. We found that inhibiting Dbr1 in mammalian cells (human neuroblastoma cell line) and in primary neurons protects against TDP-43 toxicity. Further examination of the migration of TDP-43-bound RNA with two-dimensional gels and the ability of RNA from dbr1Δ yeast to compete for TDP-43 binding in gel-mobility shift assays also supported the hypothesis that TDP-43 is binding to RNA lariats when Dbr1 is inhibited, likely contributing to the abrogation of its cellular toxicity.Citation38 Thus, the enzymatic activity of Dbr1 could be a powerful new therapeutic target for combatting ALS and other TDP-43 proteinopathies. But before Dbr1 can be leveraged as a therapeutic target in ALS, there is much more groundwork to be performed. Deeper mechanistic insight into how Dbr1 inhibition protects against TDP-43 toxicity is required as well as extension of these studies to animal models of TDP-43 proteinopathy.
We have provided evidence that the intronic lariats that accumulate in dbr1∆ cells co-localize with TDP-43 cytoplasmic inclusions. Moreover, in vitro binding assays suggest that lariats can directly compete for TDP-43 binding to RNA. Does the specific intronic lariat sequence matter or will any lariat compete for TDP-43 binding? In the future, this question can be addressed by immunoprecipitating TDP-43:RNA complexes from WT or dbr1∆ cells and performing RNA-seq to define the associated RNAs. RNase R, which degrades all cellular RNAs except for lariats, can be used to specifically enrich for lariats that associate with TDP-43 in dbr1∆ cells. It will be interesting to determine whether or not specific sequence motifs within intronic lariats are enriched or rather if specific intronic branch point architectures are enriched. If TDP-43-bound lariats can be identified, it is possible that synthetic Dbr1-resistant RNA lariats could be designed with similar sequences or structures to use as mock targets for TDP-43. This approach may specifically target rogue cytoplasmic TDP-43 without globally raising all RNA lariat species, as happens with inhibition of Dbr1.
In the future, this yeast model system could be harnessed to perform chemical screens to discover small molecule Dbr1 inhibitors. Because we have developed a methodology to visualize intronic lariats in live cells, this could be used as an easy readout to identify such chemical inhibitors. An MS2-tagged intron in a wild-type yeast strain exhibits a weak, diffuse fluorescent signal since the lariats form but are rapidly debranched by Dbr1 and then degraded. Using these yeast cells, small molecule libraries could be screened for compounds that result in the formation of fluorescent RNA lariat foci, which would suggest Dbr1 inhibition. This approach could uncover small molecules that act as full or partial inhibitors of Dbr1 function, providing a range of lariat RNA accumulation and subsequent mitigation of TDP-43 toxicity, which then could be further tested in established mammalian cell and animal models.
Our data also reveal that in yeast, when RNA lariats accumulate in the absence of Dbr1, they do so in one or two discrete foci in the cytoplasm. This suggests that 1) RNA lariats are able to get into the cytoplasm, as has been previously suggested,Citation42 and 2) that lariat RNA may be actively sequestered by the cell. It is unknown how the lariat species are getting to the cytoplasm after they are spliced from pre-mRNA in the nucleus. Again, we envision that a genetic screen using our tagged intron yeast strains could be effective at identifying the players involved in the export of lariats from the nucleus to the cytoplasm. Also, if RNA lariats are being actively sequestered in the cytoplasm along with TDP-43, it will be of interest to identify the nature of that particular cellular compartment, as its presence could be a potential target for promoting safe cytoplasmic sequestration of TDP-43. Importantly, the presence of RNA lariats in the cytoplasm observed in yeast and their interaction with TDP-43 must be confirmed in mammalian cells. Several established techniques for visualizing RNA in mammalian cells, along with Dbr1 knockdown by siRNA, can be pursued to address these issues.Citation43
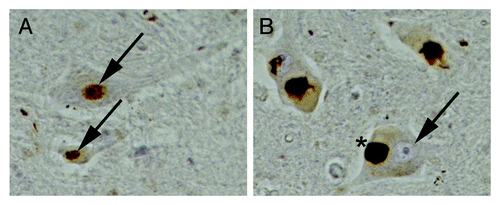
A major debate in the field is currently about how TDP-43 contributes to disease. Is it because of a loss or gain of function? exemplifies the conundrum. It shows spinal cord sections from an unaffected individual () and an ALS patient (), which we immunostained for TDP-43. The motor neurons of the control individual show diffuse nuclear staining of TDP-43. However, the motor neurons of the ALS patient show loss of TDP-43 from the nucleus and the accumulation of large round and skein-like TDP-43 aggregates in the cytoplasm. One person might look at this picture and conclude that TDP-43 is lost from the nucleus so therefore disease is caused by a loss of TDP-43’s nuclear function. But another person will look at this and say that those large cytoplasmic aggregates that accumulate must be the culprit and therefore disease is caused by a toxic gain of function in the cytoplasm. Which is right? We propose that the best evidence from the current literature suggests that both are actually right and that these two scenarios are not mutually exclusive. In other words, depletion of TDP-43 from the nucleus is obviously deleterious because of its major role as a regulator of RNA processing pathways (e.g., splicing, mRNA stability) but once it accumulates in the cytoplasm and forms aggregates, its ability to bind and sequester important RNAs and RNA-binding proteins can also have a profound effect on RNA metabolism. Therefore, strategies to deal with the loss of nuclear TDP-43 function as well as gain of toxic cytoplasmic function are needed. Our approach, especially the yeast model system, focuses on the gain of toxic function effects of TDP-43 and Dbr1 inhibition protects against cytoplasmic toxic TDP-43 aggregates. Several other approaches and model systems are being used by others to study the role of loss of TDP-43 function (e.g., knockout studies in flies, worms, fish and mice). Together, these findings will hopefully synergize and lead to a comprehensive understanding of consequences of both TDP-43 loss- and gain-of-function.
Figure 2. TDP-43 in ALS: Gain or loss-of-function? Spinal cord sections from an unaffected individual (A) or from an ALS patient (B) immunostained for TDP-43. (A) In the unaffected individual, TDP-43 is localized to the nucleus of motor neurons (arrows). (B) In motor neurons of ALS patients TDP-43 is depleted from the nucleus (arrow) and accumulates in the cytoplasm in large aggregates (*).
We have used a simple genetic model system to identify mechanisms by which TDP-43 might contribute to human ALS. Similar approaches are underway to investigate related RNA-binding proteins involved in ALS, including FUS/TLS,Citation15 and even to predict novel ALS candidate genes, such as TAF15, EWSR1, HNRNPA2B1 and HNRNPA1.Citation44-Citation46 Emerging from these studies is the concept that a large class of aggregation-prone RNA-binding proteins may contribute broadly to the pathogenesis of ALS and related neurodegenerative diseases.Citation47 It will be interesting to determine if Dbr1 inhibition can also protect against cytotoxicity from these additional disease-associated RNA-binding proteins. Finally, beyond ataxin 2 and Dbr1, our yeast TDP-43 modifier screensCitation9,Citation38(M.D.F. and A.D.G. unpublished) have revealed several additional conserved modifier genes, which promise to illuminate further disease mechanisms and hopefully point us toward better therapeutic interventions.
Acknowledgments
A.D.G. was supported by NIH Director’s New Innovator Award DP2OD004417, NIH grants R01NS065317 and R01NS073660. A.D.G. is a Pew Scholar in the Biomedical Sciences, supported by The Pew Charitable Trusts, and a Rita Allen Foundation Scholar. M.D.F. is supported by the Stanford Genome Training Program.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
References
- Andersen PM, Al-Chalabi A. Clinical genetics of amyotrophic lateral sclerosis: what do we really know?. Nat Rev Neurol 2011; 7:603 - 15; http://dx.doi.org/10.1038/nrneurol.2011.150; PMID: 21989245
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993; 362:59 - 62; http://dx.doi.org/10.1038/362059a0; PMID: 8446170
- Scott S, Kranz JE, Cole J, Lincecum JM, Thompson K, Kelly N, et al. Design, power, and interpretation of studies in the standard murine model of ALS. Amyotroph Lateral Scler 2008; 9:4 - 15; http://dx.doi.org/10.1080/17482960701856300; PMID: 18273714
- Hardiman O, van den Berg LH, Kiernan MC. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat Rev Neurol 2011; 7:639 - 49; http://dx.doi.org/10.1038/nrneurol.2011.153; PMID: 21989247
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006; 314:130 - 3; http://dx.doi.org/10.1126/science.1134108; PMID: 17023659
- Lagier-Tourenne C, Cleveland DW. Rethinking ALS: the FUS about TDP-43. Cell 2009; 136:1001 - 4; http://dx.doi.org/10.1016/j.cell.2009.03.006; PMID: 19303844
- Chen-Plotkin AS, Lee VM, Trojanowski JQ. TAR DNA-binding protein 43 in neurodegenerative disease. Nat Rev Neurol 2010; 6:211 - 20; http://dx.doi.org/10.1038/nrneurol.2010.18; PMID: 20234357
- Geser F, Martinez-Lage M, Robinson J, Uryu K, Neumann M, Brandmeir NJ, et al. Clinical and pathological continuum of multisystem TDP-43 proteinopathies. Arch Neurol 2009; 66:180 - 9; http://dx.doi.org/10.1001/archneurol.2008.558; PMID: 19204154
- Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010; 466:1069 - 75; http://dx.doi.org/10.1038/nature09320; PMID: 20740007
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011; 72:245 - 56; http://dx.doi.org/10.1016/j.neuron.2011.09.011; PMID: 21944778
- Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, et al, ITALSGEN Consortium. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011; 72:257 - 68; http://dx.doi.org/10.1016/j.neuron.2011.09.010; PMID: 21944779
- Da Cruz S, Cleveland DW. Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Curr Opin Neurobiol 2011; 21:904 - 19; http://dx.doi.org/10.1016/j.conb.2011.05.029; PMID: 21813273
- Gitler AD, Lehmann R. Modeling human disease. Science 2012; 337:269; http://dx.doi.org/10.1126/science.1227179; PMID: 22822114
- Johnson BS, McCaffery JM, Lindquist S, Gitler AD. A yeast TDP-43 proteinopathy model: Exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc Natl Acad Sci U S A 2008; 105:6439 - 44; http://dx.doi.org/10.1073/pnas.0802082105; PMID: 18434538
- Sun Z, Diaz Z, Fang X, Hart MP, Chesi A, Shorter J, et al. Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol 2011; 9:e1000614; http://dx.doi.org/10.1371/journal.pbio.1000614; PMID: 21541367
- Outeiro TF, Lindquist S. Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science 2003; 302:1772 - 5; http://dx.doi.org/10.1126/science.1090439; PMID: 14657500
- Willingham S, Outeiro TF, DeVit MJ, Lindquist SL, Muchowski PJ. Yeast genes that enhance the toxicity of a mutant huntingtin fragment or alpha-synuclein. Science 2003; 302:1769 - 72; http://dx.doi.org/10.1126/science.1090389; PMID: 14657499
- Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006; 313:324 - 8; http://dx.doi.org/10.1126/science.1129462; PMID: 16794039
- Gitler AD, Chesi A, Geddie ML, Strathearn KE, Hamamichi S, Hill KJ, et al. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat Genet 2009; 41:308 - 15; http://dx.doi.org/10.1038/ng.300; PMID: 19182805
- Krobitsch S, Lindquist S. Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc Natl Acad Sci U S A 2000; 97:1589 - 94; http://dx.doi.org/10.1073/pnas.97.4.1589; PMID: 10677504
- Giorgini F, Guidetti P, Nguyen Q, Bennett SC, Muchowski PJ. A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease. Nat Genet 2005; 37:526 - 31; http://dx.doi.org/10.1038/ng1542; PMID: 15806102
- Treusch S, Hamamichi S, Goodman JL, Matlack KE, Chung CY, Baru V, et al. Functional links between Aβ toxicity, endocytic trafficking, and Alzheimer’s disease risk factors in yeast. Science 2011; 334:1241 - 5; http://dx.doi.org/10.1126/science.1213210; PMID: 22033521
- Su LJ, Auluck PK, Outeiro TF, Yeger-Lotem E, Kritzer JA, Tardiff DF, et al. Compounds from an unbiased chemical screen reverse both ER-to-Golgi trafficking defects and mitochondrial dysfunction in Parkinson’s disease models. Dis Model Mech 2010; 3:194 - 208; http://dx.doi.org/10.1242/dmm.004267; PMID: 20038714
- Ju S, Tardiff DF, Han H, Divya K, Zhong Q, Maquat LE, et al. A yeast model of FUS/TLS-dependent cytotoxicity. PLoS Biol 2011; 9:e1001052; http://dx.doi.org/10.1371/journal.pbio.1001052; PMID: 21541368
- Johnson BS, Snead D, Lee JJ, McCaffery JM, Shorter J, Gitler AD. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J Biol Chem 2009; 284:20329 - 39; http://dx.doi.org/10.1074/jbc.M109.010264; PMID: 19465477
- Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci 2011; 14:459 - 68; http://dx.doi.org/10.1038/nn.2779; PMID: 21358643
- Barmada SJ, Skibinski G, Korb E, Rao EJ, Wu JY, Finkbeiner S. Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J Neurosci 2010; 30:639 - 49; http://dx.doi.org/10.1523/JNEUROSCI.4988-09.2010; PMID: 20071528
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008; 319:1668 - 72; http://dx.doi.org/10.1126/science.1154584; PMID: 18309045
- Zhang YJ, Xu YF, Cook C, Gendron TF, Roettges P, Link CD, et al. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc Natl Acad Sci U S A 2009; 106:7607 - 12; http://dx.doi.org/10.1073/pnas.0900688106; PMID: 19383787
- Ritson GP, Custer SK, Freibaum BD, Guinto JB, Geffel D, Moore J, et al. TDP-43 mediates degeneration in a novel Drosophila model of disease caused by mutations in VCP/p97. J Neurosci 2010; 30:7729 - 39; http://dx.doi.org/10.1523/JNEUROSCI.5894-09.2010; PMID: 20519548
- Kabashi E, Lin L, Tradewell ML, Dion PA, Bercier V, Bourgouin P, et al. Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum Mol Genet 2010; 19:671 - 83; http://dx.doi.org/10.1093/hmg/ddp534; PMID: 19959528
- Liachko NF, Guthrie CR, Kraemer BC. Phosphorylation promotes neurotoxicity in a Caenorhabditis elegans model of TDP-43 proteinopathy. J Neurosci 2010; 30:16208 - 19; http://dx.doi.org/10.1523/JNEUROSCI.2911-10.2010; PMID: 21123567
- Li Y, Ray P, Rao EJ, Shi C, Guo W, Chen X, et al. A Drosophila model for TDP-43 proteinopathy. Proc Natl Acad Sci U S A 2010; 107:3169 - 74; http://dx.doi.org/10.1073/pnas.0913602107; PMID: 20133767
- Lee T, Li YR, Chesi A, Hart MP, Ramos D, Jethava N, et al. Evaluating the prevalence of polyglutamine repeat expansions in amyotrophic lateral sclerosis. Neurology 2011; 76:2062 - 5; http://dx.doi.org/10.1212/WNL.0b013e31821f4447; PMID: 21562248
- Van Damme P, Veldink JH, van Blitterswijk M, Corveleyn A, van Vught PW, Thijs V, et al. Expanded ATXN2 CAG repeat size in ALS identifies genetic overlap between ALS and SCA2. Neurology 2011; 76:2066 - 72; http://dx.doi.org/10.1212/WNL.0b013e31821f445b; PMID: 21562247
- Ross OA, Rutherford NJ, Baker M, Soto-Ortolaza AI, Carrasquillo MM, DeJesus-Hernandez M, et al. Ataxin-2 repeat-length variation and neurodegeneration. Hum Mol Genet 2011; 20:3207 - 12; http://dx.doi.org/10.1093/hmg/ddr227; PMID: 21610160
- Fischbeck KH, Pulst SM. Amyotrophic lateral sclerosis and spinocerebellar ataxia 2. Neurology 2011; 76:2050 - 1; http://dx.doi.org/10.1212/WNL.0b013e31821f4498; PMID: 21562249
- Armakola M, Higgins MJ, Figley MD, Barmada SJ, Scarborough EA, Diaz Z, et al. Inhibition of RNA lariat debranching enzyme suppresses TDP-43 toxicity in ALS disease models. Nat Genet 2012; 44:1302 - 9; http://dx.doi.org/10.1038/ng.2434; PMID: 23104007
- Chapman KB, Boeke JD. Isolation and characterization of the gene encoding yeast debranching enzyme. Cell 1991; 65:483 - 92; http://dx.doi.org/10.1016/0092-8674(91)90466-C; PMID: 1850323
- Khalid MF, Damha MJ, Shuman S, Schwer B. Structure-function analysis of yeast RNA debranching enzyme (Dbr1), a manganese-dependent phosphodiesterase. Nucleic Acids Res 2005; 33:6349 - 60; http://dx.doi.org/10.1093/nar/gki934; PMID: 16275784
- Haim-Vilmovsky L, Gerst JE. m-TAG: a PCR-based genomic integration method to visualize the localization of specific endogenous mRNAs in vivo in yeast. Nat Protoc 2009; 4:1274 - 84; http://dx.doi.org/10.1038/nprot.2009.115; PMID: 19680241
- Hilleren PJ, Parker R. Cytoplasmic degradation of splice-defective pre-mRNAs and intermediates. Mol Cell 2003; 12:1453 - 65; http://dx.doi.org/10.1016/S1097-2765(03)00488-X; PMID: 14690599
- Weil TT, Parton RM, Davis I. Making the message clear: visualizing mRNA localization. Trends Cell Biol 2010; 20:380 - 90; http://dx.doi.org/10.1016/j.tcb.2010.03.006; PMID: 20444605
- Couthouis J, Hart MP, Shorter J, DeJesus-Hernandez M, Erion R, Oristano R, et al. A yeast functional screen predicts new candidate ALS disease genes. Proc Natl Acad Sci U S A 2011; 108:20881 - 90; http://dx.doi.org/10.1073/pnas.1109434108; PMID: 22065782
- Couthouis J, Hart MP, Erion R, King OD, Diaz Z, Nakaya T, et al. Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis. Hum Mol Genet 2012; 21:2899 - 911; http://dx.doi.org/10.1093/hmg/dds116; PMID: 22454397
- Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013; 495:467 - 73; http://dx.doi.org/10.1038/nature11922; PMID: 23455423
- King OD, Gitler AD, Shorter J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res 2012; 1462:61 - 80; http://dx.doi.org/10.1016/j.brainres.2012.01.016; PMID: 22445064