Abstract
Hereditary sensory and autonomic neuropathy type 2 is a rare autosomal recessive pathology presenting with early onset peripheral sensory defects. It arises from mutations affecting a specific isoform of the WNK1 kinase (with-no-lysine protein kinase 1) termed WNK1/HSN2. The role of WNK1 in the nervous system is not well understood. In our recent paper, we examined the effect of a pathological loss-of-function of the Wnk1/Hsn2 isoform on the development of the peripheral nervous system of the zebrafish embryo. Upon Wnk1/Hsn2 silencing using antisense morpholino oligonucleotides, we observed defects in the development of the sensory peripheral lateral line (PLL). Phenotypical embryos were also found to overexpress RNA for potassium-chloride cotransporter 2 (KCC2), a downstream target of WNK1 phosphorylation. Injection of recombinant mRNA for active KCC2, but not for inactive mutant KCC2-C568A, replicated the PLL defects observed in wnk1/hsn2 deficient animals, suggesting an essential role for WNK1/HSN2 in KCC2 regulation.
Hereditary sensory and autonomic neuropathy type 2 (HSANII) is an early onset peripheral neuropathy characterized by lack of sensory perception for all modalities in the distal limbs. Early evidence from sural nerve biopsies of HSANII patients showed a lower number of sensory fibers but no sign of neurodegeneration, leading to the hypothesis that this pathology has a developmental originCitation1,Citation2.
Many mutations have been identified in HSANII patients, most of which are found in the alternatively spliced “HSN2” exon of the WNK1 gene (with-no-lysine protein kinase 1), coding for a ubiquitously expressed serine/threonine kinase. The WNK1/HSN2 isoform has a marked expression in the nervous system and a specific axonal localization at the level of the neuron.Citation3,Citation4 HSANII-linked mutations are located within the HSN2 exon and lead to a truncated protein isoform, which is non-functional. As WNK kinases (four family members) are known to regulate the activity of potassium-chloride cotransporters of the KCC family (coded by SLC12 genes) by phosphorylation,Citation5 our recent paper focused on the role of protein isoform WNK1/HSN2 and its interaction with KCC2 (Potassium-chloride cotransporter member 5, SLC125A or KCC2 gene) in HSANII pathogenesis.
The posterior lateral line (PLL) of the zebrafish embryo appeared to be an appropriate model for studying the role of Wnk1/Hsn2 in peripheral nerve development. Indeed, we observed expression of Wnk1/Hsn2 and Kcc2 in the neuromasts of the PLL, a peripheral mechanosensory system formed when a group of progenitor cells (the primordium) migrates from the cephalic placodes along the midline of the tail and deposits bundles of cells, leaving behind the PLL ganglion. The progenitor bundles then differentiate into functional sensory organs named neuromasts, using Delta/Notch signaling to specify differentiation into either support cells or sensory hair cells. The posterior lateral line nerve, trailing the migration of the primordium, will innervate the neuromasts, completing a basic functional PLL by 72 h post-fertilization (hpf).Citation6,Citation7
Using antisense oligonucleotide technology, we silenced the expression of the Wnk1/Hsn2 isoform in embryos to mimic the pathogenic loss of function in HSANII and observed a defect in the development of the PLL. The knockdown embryos had fewer neuromast organs, which were themselves composed of fewer cells. The PLL progenitor primordium was also of a smaller size in absence of Wnk1/Hsn2, despite being constituted of normal sized and adequately organized cells. These defects could be partially rescued by the injection of a recombinant human WNK1 mRNA, but not by injection of mRNA coding for a short variant mimicking the truncation of the protein resulting from HSNAII mutations, even though both constructs contained the full HSN2 exon.
Previous studies showed that WNK1 is responsible for phosphorylating sites that control the cotransporter activity in all KCC family members,Citation5 probably by inhibiting surface expression at the cell membrane.Citation8 It was found that phosphorylation by WNK1 is essential for deactivation of the KCCs and that it occurs in immature neurons but is absent in adult neuronsCitation5,Citation9. This is consistent with the expression pattern described in the zebrafish embryo, where kcc2 is increasingly expressed as neurons mature.Citation10 KCC2 was also reported as having a rapid turnover rate at the cell membrane, suggesting it has the capacity to respond quickly to changes in the environment. Additionally, KCC2 activation has been shown to be influenced by neuronal activity, neuronal, and oxidative stress, and to regulate transcription and expression levels of the co-transporter, a phenomenon possibly dependent on phosphorylation.Citation11,Citation12 Since we also observed wnk1/hsn2 expression during early development, we postulated that the regulation of KCC2 by WNK1 might be affected by HSANII mutations, where the loss of function of the kinase isoform would lead to a lack of phosphorylation of the cotransporter, necessary for its deactivation, and possibly, for its downregulation.
We therefore looked at the expression levels of the zebrafish KCC2 ortholog kcc2 (gene slc12a5) and found an overexpression at the mRNA level in Wnk1/Hsn2 knockdown embryos at 72hpf, when the developmental phenotype is visible, but also at 24hpf, when the primordium has just begun its migration. To test whether the PLL defects observed were due to KCC2 overexpression, we performed a controlled overexpression by injection of recombinant KCC2 mRNA in wild-type zebrafish eggs, producing embryos phenocopying Wnk1/Hsn2 silencing. These results suggest that KCC2 overexpression might be downstream of WNK1/HSN2 in HSANII pathogenesis. To further validate this new pathogenic pathway, we performed a double knockdown experiment where sub-optimal doses of AMOs were used in combination, to partially knockdown the expression of both Wnk1/Hsn2 and Kcc2. While each AMO injected by itself produced no discernible phenotype, the additive effect of the co-injection phenocopied the PLL defects observed upon Wnk1/Hsn2 silencing.
To determine if the defects observed in the PLL development following KCC2 mRNA overexpression were due to the cotransporter’s function at the membrane, we injected recombinant mRNA for an inactive KCC2 point mutant, KCC2-C568A. This dominant negative mutant was shown to lead to disrupted chloride extruding function and to be incapable of being expressed at the cell membrane due to impaired interaction with structural protein 4.1N, responsible for linking the cotransporter with the cytoskeleton.Citation10,Citation14 We observed that an overexpression of KCC2-C568A mRNA mimicked the overexpression of wild-type KCC2 mRNA and the knockdown of Wnk1/Hsn2 by producing embryos with the previously described PLL defects. We therefore propose that WNK1 mutations linked with HSANII function via an overexpression of KCC2 mRNA, and that the cotransporter can act independently of its chloride-extruding function to interfere with proper development of the peripheral sensory system (). This is consistent with recent reports that KCC2 in developing rat brain is found in a larger proportion in the cytoplasm, rather than being associated with the plasma membrane, supporting a role independent of its chloride-extruding function.Citation13
Figure 1. Model of the proposed mechanism involving regulation of KCC2 activation and transcription, both by levels of activated KCC2 and by levels of KCC2 protein with or without phosphorylation by WNK1/HSN2.
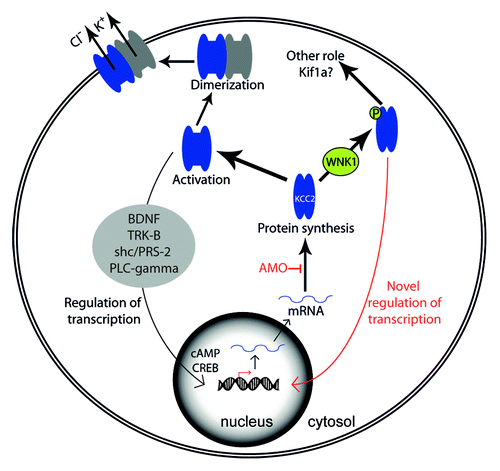
The expression of this neural-specific cotransporter is responsible for the developmental GABAergic response switch. Effectively, in the mature brain, GABA is the main inhibitory neurotransmitter, but it is excitatory during development. In immature neurons, KCC2 is absent from the membrane, but antagonistic co-transporter NKCC1 is active, leading to a higher intracellular chloride concentration. The activation of KCC2 takes place when neurons undergo maturation and once it is located at the membrane, causes the efflux of K-Cl from the cell, leading to a reduced intracellular chloride concentration. This results in a hyperpolarizing (inhibitory) response to GABA and glycine, where it was excitatory during early development.Citation15 The presence and activation of KCC2 is therefore narrowly controlled in time as proper neurogenesis depends on its chloride extrusion function. In the zebrafish embryo, neurogenesis in the spinal cord has been shown to be regulated by Kcc2, where a precocious overexpression of the cotransporter led to fewer neurons, stalling progenitors before differentiation.Citation10 At the level of the PLL, we found that neuromast hair cells loaded with a Ca2+ indicator did not respond to glycine application but evoked Ca2+ transients upon glutamate application, suggesting that these cells are viable but that the presence of Kcc2, responsible for lowering intracellular chloride levels, made them unresponsive to glycine. On the contrary, progenitor cells composing the PLL primordium responded to the application of glycine. These results suggest that the PLL progenitors, like the ones found in the spinal cordCitation10 have a high intracellular chloride concentration and support the hypothesis that Kcc2 expression at the level of the neuromasts might have a role in their differentiation.
Although phosphorylation studies were not feasible in our model, it would be of great interest to understand the interaction between KCC2 and WNK1. It was previously suggested that regulation of KCC2 activation by the WNK kinases might be a direct interaction, or via the inactivation of downstream phosphatases, but it might also implicate the SPAK/OSR1 pathway, as is the case for phosphorylation of antagonist co-transporter NKCC1.Citation16 However, it was reported that the expression of a mutant form of WNK3 lacking a specific SPAK binding site (WNK3-FI337A) led to loss of activation for NKCC2 but retained inhibition of KCC4 activity, suggesting the latter to be independent of SPAK/OSR1 interaction.Citation17 Additionally, knockdown of SPAK and OSR1 did not affect phosphorylation of sites T991 and T1048 of KCC3, while knockdown of WNK1, but not other WNK family members, reduced it.Citation5 These results suggest that while the regulation of KCC and NKCC are both dependent on WNK1 activity, only NKCCs are phosphorylated via downstream kinases SPAK/OSR1. As our in vivo model did not permit verification of the ratio of plasmalemmal and cytosolic KCC2 expression or its phosphorylation state in either localization, further study needs to be done to uncover more of the pathogenic pathway leading to HSANII-linked developmental deficits.
Recent studies describe the presence of the WNK1/HSN2 isoform outside of the nervous system, where it was thought to be confined.Citation5,Citation18 While presence of this isoform in non-neuronal tissue, like kidneys and testis might not be explained by our HSANII pathogenesis model, presence in satellite cells, which are glial cells of the peripheral nervous system,Citation18 could also support the involvement of KCC2 in HSANII pathogenesis. Indeed, KCC2 was found to be expressed at the level of the nodal segments of peripheral nerve axons and in Schwann cells,Citation19 and to co-localize with transport vesiclesCitation13 indicating an overlap with the localization of the WNK1/HSN2 isoform. In fact, early reports showed that this isoform has a particular sub-localization within neurons when compared with other WNK1 isoforms: it is also expressed at the level of the axons while others are restricted to the cell body.Citation3 As KIF1a (coding for kinesin family member 3) mutations have been found in HSANII patients,Citation20 it is tempting to speculate that the WNK1/HSN2 isoform has other functions that are essential to the proper development of peripheral sensory neurons. Indeed, the KIF1a kinesin, a motor protein responsible for anterograde axonal transport of synaptic vesicles, was reported to interact with a domain encoded by the HSN2 exon.Citation20 As WNK1/HSN2 and Kif1a co-localize at the level of the axon, we could hypothesize that this specific interaction affects the function of the motor protein, whether it be via phosphorylation, which has been shown to influence both cargo binding and motor domain stability, or through other mechanisms.Citation21,Citation22 In the context of WNK1/HSN2 mutations, we could conjecture that the few peripheral sensory neurons that would develop following pathogenic changes in KCC2 activity and expression during early development would do so improperly, perhaps due to deficient axonal transport. The resulting alterations, possibly in distribution of specific cargo at the nodes of Ranvier, could then interfere with their function.
While our model is a first insight into the mechanisms underlying HSANII pathophysiology, much work remains to be done to uncover the functional contribution of the WNK1/HSN2 kinase isoform to health and disease as it has proven to have an essential role in proper development of the peripheral sensory system.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Acknowledgments
VB is recipient of the ENP Graduate Program fellowship from École des Neurosciences de Paris Ile-de-France and the Fonds de Recherche en Santé du Québec (FRSQ) doctoral award. The original research was supported by the MD Foundation, Natural Sciences and Engineering Research Council of Canada (NSERC) and the Canadian Institutes of Health Research (CIHR).
References
- Murray TJ. Congenital sensory neuropathy. Brain 1973; 96:387 - 94; http://dx.doi.org/10.1093/brain/96.2.387; PMID: 4123669
- Axelrod FB, Gold-von Simson G. Hereditary sensory and autonomic neuropathies: types II, III, and IV. Orphanet J Rare Dis 2007; 2:39; http://dx.doi.org/10.1186/1750-1172-2-39; PMID: 17915006
- Shekarabi M, Girard N, Rivière JB, Dion P, Houle M, Toulouse A, Lafrenière RG, Vercauteren F, Hince P, Laganiere J, et al. Mutations in the nervous system--specific HSN2 exon of WNK1 cause hereditary sensory neuropathy type II. J Clin Invest 2008; 118:2496 - 505; PMID: 18521183
- Vidal-Petiot E, Cheval L, Faugeroux J, Malard T, Doucet A, Jeunemaitre X, Hadchouel J. A new methodology for quantification of alternatively spliced exons reveals a highly tissue-specific expression pattern of WNK1 isoforms. PLoS One 2012; 7:e37751; http://dx.doi.org/10.1371/journal.pone.0037751; PMID: 22701532
- Rinehart J, Maksimova YD, Tanis JE, Stone KL, Hodson CA, Zhang J, Risinger M, Pan W, Wu D, Colangelo CM, et al. Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell 2009; 138:525 - 36; http://dx.doi.org/10.1016/j.cell.2009.05.031; PMID: 19665974
- Ghysen A, Dambly-Chaudière C. Le développement du système nerveux: de la mouche au poisson, du poisson à l’homme... Med Sci (Paris) 2003; 19:575 - 81; http://dx.doi.org/10.1051/medsci/2003195575; PMID: 12836391
- Ghysen A, Dambly-Chaudière C. The lateral line microcosmos. Genes Dev 2007; 21:2118 - 30; http://dx.doi.org/10.1101/gad.1568407; PMID: 17785522
- Rinehart J, Vázquez N, Kahle KT, Hodson CA, Ring AM, Gulcicek EE, Louvi A, Bobadilla NA, Gamba G, Lifton RP. WNK2 kinase is a novel regulator of essential neuronal cation-chloride cotransporters. J Biol Chem 2011; 286:30171 - 80; http://dx.doi.org/10.1074/jbc.M111.222893; PMID: 21733846
- Kahle KT, Rinehart J, de Los Heros P, Louvi A, Meade P, Vazquez N, Hebert SC, Gamba G, Gimenez I, Lifton RP. WNK3 modulates transport of Cl- in and out of cells: implications for control of cell volume and neuronal excitability. Proc Natl Acad Sci U S A 2005; 102:16783 - 8; http://dx.doi.org/10.1073/pnas.0508307102; PMID: 16275911
- Reynolds A, Brustein E, Liao M, Mercado A, Babilonia E, Mount DB, Drapeau P. Neurogenic role of the depolarizing chloride gradient revealed by global overexpression of KCC2 from the onset of development. J Neurosci 2008; 28:1588 - 97; http://dx.doi.org/10.1523/JNEUROSCI.3791-07.2008; PMID: 18272680
- Wake H, Watanabe M, Moorhouse AJ, Kanematsu T, Horibe S, Matsukawa N, Asai K, Ojika K, Hirata M, Nabekura J. Early changes in KCC2 phosphorylation in response to neuronal stress result in functional downregulation. J Neurosci 2007; 27:1642 - 50; http://dx.doi.org/10.1523/JNEUROSCI.3104-06.2007; PMID: 17301172
- Rivera C, Voipio J, Thomas-Crusells J, Li H, Emri Z, Sipilä S, Payne JA, Minichiello L, Saarma M, Kaila K. Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J Neurosci 2004; 24:4683 - 91; http://dx.doi.org/10.1523/JNEUROSCI.5265-03.2004; PMID: 15140939
- Kovács K, Basu K, Rouiller I, Sík A. Regional differences in the expression of K(+)-Cl (-) 2 cotransporter in the developing rat cortex. Brain Struct Funct 2013; http://dx.doi.org/10.1007/s00429-013-0515-9; PMID: 23420348
- Horn Z, Ringstedt T, Blaesse P, Kaila K, Herlenius E. Premature expression of KCC2 in embryonic mice perturbs neural development by an ion transport-independent mechanism. Eur J Neurosci 2010; 31:2142 - 55; http://dx.doi.org/10.1111/j.1460-9568.2010.07258.x; PMID: 20529123
- Brustein E, Drapeau P. Serotoninergic modulation of chloride homeostasis during maturation of the locomotor network in zebrafish. J Neurosci 2005; 25:10607 - 16; http://dx.doi.org/10.1523/JNEUROSCI.2017-05.2005; PMID: 16291933
- Kahle KT, Rinehart J, Lifton RP. Phosphoregulation of the Na-K-2Cl and K-Cl cotransporters by the WNK kinases. Biochim Biophys Acta 2010; 1802:1150 - 8; http://dx.doi.org/10.1016/j.bbadis.2010.07.009; PMID: 20637866
- Pacheco-Cuellar G, González-Huerta LM, Valdés-Miranda JM, Peláez-González H, Zenteno-Bacheron S, Cazarin-Barrientos J, Cuevas-Covarrubias SA. Hereditary sensory and autonomic neuropathy II due to novel mutation in the HSN2 gene in Mexican families. J Neurol 2011; 258:1890 - 2; http://dx.doi.org/10.1007/s00415-011-6025-x; PMID: 21625937
- Shekarabi M, Lafrenière RG, Gaudet R, Laganière J, Marcinkiewicz MM, Dion PA, Rouleau GA. Comparative analysis of the expression profile of Wnk1 and Wnk1/Hsn2 splice variants in developing and adult mouse tissues. PLoS One 2013; 8:e57807; http://dx.doi.org/10.1371/journal.pone.0057807; PMID: 23451271
- Sun Y-T, Lin T-S, Tzeng S-F, Delpire E, Shen M-R. Deficiency of electroneutral K+-Cl- cotransporter 3 causes a disruption in impulse propagation along peripheral nerves. Glia 2010; 58:1544 - 52; PMID: 20549748
- Rivière J-B, Ramalingam S, Lavastre V, Shekarabi M, Holbert S, Lafontaine J, Srour M, Merner N, Rochefort D, Hince P, et al. KIF1A, an axonal transporter of synaptic vesicles, is mutated in hereditary sensory and autonomic neuropathy type 2. Am J Hum Genet 2011; 89:219 - 30; http://dx.doi.org/10.1016/j.ajhg.2011.06.013; PMID: 21820098
- Sato-Yoshitake R, Yorifuji H, Inagaki M, Hirokawa N. The phosphorylation of kinesin regulates its binding to synaptic vesicles. J Biol Chem 1992; 267:23930 - 6; PMID: 1429730
- Morfini G, Pigino G, Szebenyi G, You Y, Pollema S, Brady ST. JNK mediates pathogenic effects of polyglutamine-expanded androgen receptor on fast axonal transport. Nat Neurosci 2006; 9:907 - 16; http://dx.doi.org/10.1038/nn1717; PMID: 16751763