Abstract
The gray platelet syndrome (GPS) is a rare, autosomal-recessive platelet disorder characterized by thrombocytopenia, large platelets lacking α-granules, and variable bleeding. GPS has been linked to mutations in the neurobeachin-like 2 gene (NBEAL2). We have recently characterized Nbeal2-deficient mice and shown that the absence of Nbeal2 results in defective protein sorting in megakaryocytes (MKs) and impaired α-granule biogenesis, a finding also seen for human MKs. In the mice, the lack of α-granules results in impaired aggregation, defective platelet adhesion to collagen under flow and reduced pro-coagulant activity; findings that translate into defective hemostasis and thrombosis in vivo indicating that α-granule secretion is critical for platelet plug stability. Furthermore, we revealed a role of α-granule proteins in ischemic stroke and wound healing. Thus, Nbeal2-deficient mice recapitulate the hallmarks of human GPS without showing its phenotypic heterogeneity and are a promising model to investigate the (patho-)physiological relevancy of α-granules.
Keywords: :
The Gray Platelet Syndrome
The gray platelet syndrome (GPS) is a rare inherited platelet disorder that is characterized by mild to moderate thrombocytopenia with large platelets that lack α-granules.Citation1,Citation2 Platelets are small anuclear cell fragments that safeguard vessel wall integrity. Vessel damage exposes the subendothelial extracellular matrix (ECM) and induces rapid deceleration of circulating platelets, thereby enabling sustained contacts of platelet receptors with ECM proteins resulting in platelet activation.Citation3 This activation causes a rapid remodeling of the cytoskeleton and a morphological change of the platelets from discoid to spheroid followed by spreading on the reactive surface. Platelet activation results in the exocytosis of α- and dense granules, small intracellular organelles that are exclusive to platelets and their progenitors, megakaryocytes. While dense granules contain non-proteinaceous compounds such as calcium, serotonin, ADP and ATP that foster platelet aggregation, α-granules contain a plethora of more than 300 different proteins involved not only in platelet adhesion but also inflammation, angiogenesis and wound healing.Citation4
Platelet plug formation is critical to seal the wound site preventing blood loss and building a first line of defense against pathogens that otherwise might enter the circulation. Platelets also contribute to wound repair and tissue regeneration. In cardiovascular disease, platelet activation on atherosclerotic plaques can lead to excessive thrombus formation, which may cause vessel occlusion, infarction and death.Citation5
GPS was initially described in 1971,Citation6 based on the observation that a May–Grünwald-Giemsa-stained blood smear of a patient with moderate bleeding showed enlarged platelets that appeared gray. Further studies revealed lack of α-granules and their contents in platelets and MKs.Citation7,Citation8 This results in platelet aggregation defects, myelofibrosis, splenomegaly and mild to moderate or even severe bleeding complications in human patients.Citation1,Citation2 Recently, the gene that is mutated in GPS was identified as Neurobeachin-like 2 (NBEAL2).Citation9-Citation11 NBEAL2 encodes a 302 kDa member of the family of BEACH (Beige and Chediak-Higashi) domain-containing proteins, which are believed to be important in membrane protein trafficking.Citation12
Nbeal2-Deficient Mice
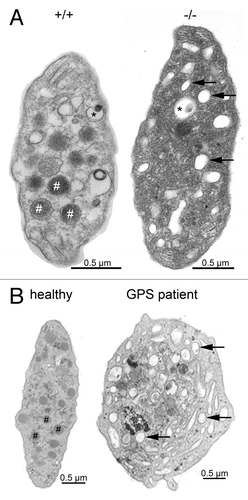
Using a reverse genetic approach we recently showed that Nbeal2-deficient mice mimic most of the important hallmarks of GPS thereby establishing this mouse as a model of the disease,Citation13 a finding that was confirmed by a second group.Citation14 As in human GPS, Nbeal2-deficient mice exhibit thrombocytopenia with enlarged platelets, a mild splenomegaly and absence of α-granules in platelets. While taking into account the smaller size of mouse platelets, Nbeal2−/− platelets show the same morphological characteristics as human GPS including a high number of vacuoles, which mostly appear empty (). The content of α-granule proteins synthesized by MKs, such as von Willebrand factor (VWF), platelet factor 4 (PF4) and thrombospondin-1 was strongly reduced in the knockout platelets.Citation13,Citation14 The platelet fibrinogen content was also decreased, indicating that endocytosed proteins also fail to be stored in Nbeal2−/− platelets, as in the human disease. On the other hand, P-selectin, which serves widely as a marker for α-granule secretion and is a component of the α-granule membrane, was still present, albeit at lower levels; its surface exposure upon activation reached approximately 50% of wild-type levels. For human GPS, P-selectin has been shown to be either reduced or normal.Citation15 In line with observations for GPS patients,Citation2 dense granule secretion was not affected in Nbeal2−/− platelets.Citation13,Citation14
Figure 1. Platelet ultrastructure in Nbeal2−/− mice (A) and human GPS (B). Representative transmission electron microscopy (TEM) images of resting wild-type (+/+) and Nbeal2−/− (−/−) platelets and those of a control human subject as well as a characterized GPS patientCitation9,Citation15 with a homozygous L388P mutation. Platelets deficient in NBEAL2 both show lack of α-granules (#) and an increased number of vacuoles (arrows) while platelet size was increased and dense granule (*) content was unaltered. Bars = 0.5 µm.
As the knockout mice showed a thrombocytopenia with platelet counts reduced by about 40% we further investigated megakaryocytopoiesis and found that Nbeal2-deficient megakaryocytes (MKs) were generally able to produce proplatelets, showed frequent emperipolesis (the presence of leukocytes within the cytoplasm) and lacked α-granules. Instead, MKs contained many vacuoles. Interestingly, we found VWF mislocalization in mature MKs with either accumulation in one part of the MK or complete loss of the protein. This indicates that protein synthesis per se might not be affected. Moreover, platelet lifespan within the circulation was not altered by the absence of Nbeal2.Citation13 Kahr et al., taking a different approach, reported that Nbeal2-deficient MKs have a less developed proplatelet territory and are stalled at early developmental stages when cultured in the presence of thrombopoietin.Citation14 As we did not observe such severe defects in the proplatelet territories and ploidy of Nbeal2-deficient MKs taken from bone marrow,Citation13 further studies will be needed to clarify these differences.
Functional Consequences of Nbeal2-Deficiency in Vivo
The production of the Nbeal2−/− mouse has for the first time allowed an evaluation of the in vivo significance of platelet α-granule deficiency. A combination of impaired aggregation, defective platelet adhesion to collagen under flow and reduced pro-coagulant activity was associated with prolonged tail bleeding times and the mice were protected in in vivo models of arteriolar thrombosis and ischemic stroke. Of note, these findings are most likely caused by the functional platelet defect and not by the thrombocytopenia, since platelet count reductions of up to 70% were without effect in these models.Citation16 Interestingly, Nbeal2−/− mice did not display signs of spontaneous bleeding, nor did we observe intracranial hemorrhage within 24 h after cerebral ischemia. This indicates that α-granule proteins might be dispensable for maintaining vascular integrity under basal or thrombo-inflammatory conditions. On the other hand, upon injury, α-granule secretion is required to prevent excessive blood loss, as shown by prolonged tail bleeding times. Moreover, we demonstrated a role for platelet α-granule secretion in wound healing, since Nbeal2−/− mice displayed reduced collagenous granulation tissue formation, indicative of a reduced myofibroblast infiltration.Citation13 Experiments with bone marrow chimeric mice further demonstrated that Nbeal2 expression in the hematopoietic system is required for normal wound healing (not shown), prevention of excessive blood loss and for the formation of stable vessel occluding thrombi in vivo.Citation13
Nbeal2-Deficient Mice Mimic Human GPS
Nbeal2−/− mice recapitulate the hallmarks of GPS patients with the absence of α-granules and their content. Nevertheless, a few differences exist. We did not observe the heterogeneity of the platelet aggregation responses seen in GPS patients upon stimulation with different agonists.Citation1,Citation2 This could be explained by the different mutations that were found in the NBEAL2 gene, which in contrast to the full knockout in the mouse model, may not necessarily lead to a complete loss of protein function or expression.Citation9-Citation11,Citation17 Furthermore, Nbeal2−/− mice all have a common genetic background while in human disease phenotype will be influenced by the whole range of single-nucleotide polymorphisms that define the hemostatic response. Similarly, platelet count is highly variable in GPS patients and thrombocytopenia can be severe, which almost certainly contributes to bleeding. Another frequent characteristic of GPS patients is myelofibrosis probably caused by α-granule proteins being released from MKs into the marrow with deregulated cell proliferation and differentiation. In the mice, signs of myelofibrosis were not observed in young as well as in 4- and 6-mo-old animals.Citation13,Citation14 Whether onset of myelofibrosis will occur later in these animals needs to be investigated.
Notwithstanding these differences, we believe that Nbeal2−/− mice represent an important model for GPS and that they will help our understanding of the physiologic role of the α-granule pool of proteins. Importantly, Nbeal2−/− mice will enable studies on the effects of Nbeal2-deficiency in other cell types. While Nbeal2 expression levels in other hematopoietic cells are low compared with MKs,Citation9,Citation13 our preliminary studies have revealed functional consequences of Nbeal2-deficiency in immune cells (unpublished observations). Also of special interest will be a potential relevance of α-granule content in mediating lymph-vessel separationCitation18 and vascular integrity upon inflammation.Citation19,Citation20 Both processes depend on platelets but apparently do not require integrin-activation indicating that platelet granule secretion might be the critical step.
Mechanistical Function of NBEAL2
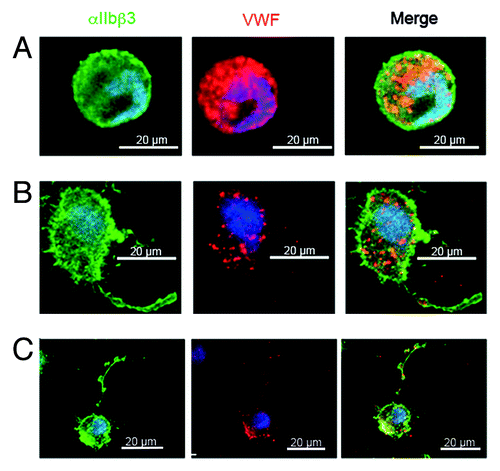
Beyond the functional analyses, the question of the mechanism by which NBEAL2 regulates protein sorting during α-granule biogenesis, remains to be addressed. NBEAL2 is a large, multi-domain protein that consists of 2754 amino acids and contains armadillo-like (ARM), concanavalin-A-like (ConA-like), pleckstrin homology (PH) and BEACH and WD40 (tryptophan-aspartic acid repeat containing) domains. Of note, GPS-causing point mutations are not limited to one particular domain but are distributed throughout the entire protein.Citation9-Citation11,Citation17 While it is thought that ConA-like lectin domains could be involved in protein sorting and secretion,Citation21 it is known that WD40 domains are involved in protein-protein interactions and PH domains facilitate membrane association. BEACH domain containing proteins are of emerging clinical importance, as more members of this family become associated with human disease.Citation22 However, they act in different cellular processes including vesicular transport, apoptosis, membrane dynamics and receptor signaling. For NBEAL2 a role in membrane dynamics and vesicular transport is consistent with the finding that NBEAL2-deficiency causes a dramatically increased number of empty vacuoles and mistargeting of α-granule proteins that leads to loss or accumulation of the protein. This is also shown for human GPS, a finding illustrated in where MKs cultured in vitro from peripheral blood CD34+ cells from a typical GPS patient (with a homozygous L388P mutation),Citation9 clearly show synthesis of VWF but with loss prior to proplatelet formation and a lack of VWF trafficking along proplatelets in mature cells. It seems therefore that NBEAL2 acts at an early stage of α-granule biogenesis as do other previously identified regulators of α-granule biogenesis VPS33B and VPS16B.Citation23 Significantly, protein levels of these two proteins were unchanged in Nbeal2−/− platelets.Citation14
Figure 2. Confocal microscopy of MKs cultured in vitro from CD34+ cells from the peripheral blood of a GPS patient with a homozygous L388P mutation in NBEAL2,Citation9 MK suspensions were incubated at day 14 of culture on polylysine-coated slides. Cells were fixed and permeabilized. Integrin αIIbβ3 and VWF were localized by a murine antibody (AP2) and VWF by a polyclonal antibody with bound IgG visualized using species-specific FITC (green) and Alexa-Fluor568-conjugated IgG. All technical details were as previously described.Citation24 In the upper panel, a round small MK shows abundant labeling for VWF; in the middle panel a MK shows VWF staining along the proplatelet extension, the VWF labeling is decreased compared to control and is absent from the proplatelet tip; the lower panel shows a very mature MK with a long proplatelet string with typical swellings along its length while the VWF labeling is minimal. This suggests that VWF is not maintained in the MK. Please note that the scale of this panel is reduced to allow the proplatelet to be fully seen.
Conclusions
Our study on Nbeal2−/− mice confirms that the absence of Nbeal2 results in defective protein sorting in MKs and impaired α-granule biogenesis. Thus, Nbeal2-deficient mice recapitulate many of the hallmarks of GPS patients and can therefore be used to study the in vivo consequences of α-granule deficiency as well as studying potential therapeutic options in the treatment of GPS. Using this mouse model we have provided experimental evidence of the importance of α-granule proteins in thrombosis, ischemic stroke and wound healing. Future studies will address the relevance of NBEAL2 in other cell types and will attempt to elucidate how NBEAL2 facilitates α-granule biogenesis.
| Abbreviations: | ||
| ARM | = | armadillo-like domain |
| BEACH | = | Beige and Chediak-Higashi |
| ConA | = | concanavalin-A |
| ECM | = | extracellular matrix |
| GPS | = | gray platelet syndrome |
| MK | = | megakaryocyte |
| Nbeal2 | = | Neurobeachin-like 2 |
| PF4 | = | platelet-factor 4 |
| PH | = | Pleckstrin homology |
| VWF | = | von Willebrand factor |
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Acknowledgments
We acknowledge the contributions of all the co-authors of our recent JCI publication. This work was supported by the Deutsche Forschungsgemeinschaft (SFB 688) and the Rudolf Virchow Center. CD was supported by a grant of the German Excellence Initiative to the Graduate School of Life Sciences, University of Würzburg.
References
- Gunay-Aygun M, Zivony-Elboum Y, Gumruk F, Geiger D, Cetin M, Khayat M, Kleta R, Kfir N, Anikster Y, Chezar J, et al. Gray platelet syndrome: natural history of a large patient cohort and locus assignment to chromosome 3p. Blood 2010; 116:4990 - 5001; http://dx.doi.org/10.1182/blood-2010-05-286534; PMID: 20709904
- Nurden AT, Nurden P. The gray platelet syndrome: clinical spectrum of the disease. Blood Rev 2007; 21:21 - 36; http://dx.doi.org/10.1016/j.blre.2005.12.003; PMID: 16442192
- Stegner D, Nieswandt B. Platelet receptor signaling in thrombus formation. J Mol Med (Berl) 2011; 89:109 - 21; http://dx.doi.org/10.1007/s00109-010-0691-5; PMID: 21058007
- Blair P, Flaumenhaft R. Platelet alpha-granules: basic biology and clinical correlates. Blood Rev 2009; 23:177 - 89; http://dx.doi.org/10.1016/j.blre.2009.04.001; PMID: 19450911
- Jackson SP. Arterial thrombosis--insidious, unpredictable and deadly. Nat Med 2011; 17:1423 - 36; http://dx.doi.org/10.1038/nm.2515; PMID: 22064432
- Raccuglia G. Gray platelet syndrome. A variety of qualitative platelet disorder. Am J Med 1971; 51:818 - 28; http://dx.doi.org/10.1016/0002-9343(71)90311-1; PMID: 5129551
- Breton-Gorius J, Vainchenker W, Nurden A, Levy-Toledano S, Caen J. Defective alpha-granule production in megakaryocytes from gray platelet syndrome: ultrastructural studies of bone marrow cells and megakaryocytes growing in culture from blood precursors. Am J Pathol 1981; 102:10 - 9; PMID: 7468753
- Rosa JP, George JN, Bainton DF, Nurden AT, Caen JP, McEver RP. Gray platelet syndrome. Demonstration of alpha granule membranes that can fuse with the cell surface. J Clin Invest 1987; 80:1138 - 46; http://dx.doi.org/10.1172/JCI113171; PMID: 2443536
- Albers CA, Cvejic A, Favier R, Bouwmans EE, Alessi M-C, Bertone P, Jordan G, Kettleborough RN, Kiddle G, Kostadima M, et al. Exome sequencing identifies NBEAL2 as the causative gene for gray platelet syndrome. Nat Genet 2011; 43:735 - 7; http://dx.doi.org/10.1038/ng.885; PMID: 21765411
- Gunay-Aygun M, Falik-Zaccai TC, Vilboux T, Zivony-Elboum Y, Gumruk F, Cetin M, Khayat M, Boerkoel CF, Kfir N, Huang Y, et al. NBEAL2 is mutated in gray platelet syndrome and is required for biogenesis of platelet α-granules. Nat Genet 2011; 43:732 - 4; http://dx.doi.org/10.1038/ng.883; PMID: 21765412
- Kahr WH, Hinckley J, Li L, Schwertz H, Christensen H, Rowley JW, Pluthero FG, Urban D, Fabbro S, Nixon B, et al. Mutations in NBEAL2, encoding a BEACH protein, cause gray platelet syndrome. Nat Genet 2011; 43:738 - 40; http://dx.doi.org/10.1038/ng.884; PMID: 21765413
- Wang X, Herberg FW, Laue MM, Wullner C, Hu B, Petrasch-Parwez E, Kilimann MW. Neurobeachin: A protein kinase A-anchoring, beige/Chediak-higashi protein homolog implicated in neuronal membrane traffic. J Neurosci 2000; 20:8551 - 65; PMID: 11102458
- Deppermann C, Cherpokova D, Nurden P, Schulz JN, Thielmann I, Kraft P, Vögtle T, Kleinschnitz C, Dütting S, Krohne G, et al. Gray platelet syndrome and defective thrombo-inflammation in Nbeal2-deficient mice. J Clin Invest 2013; 123:3331 - 42; http://dx.doi.org/10.1172/JCI69210; PMID: 23863626
- Kahr WH, Lo RW, Li L, Pluthero FG, Christensen H, Ni R, Vaezzadeh N, Hawkins CE, Weyrich AS, Di Paola J, et al. Abnormal megakaryocyte development and platelet function in Nbeal2-/- mice. Blood 2013; http://dx.doi.org/10.1182/blood-2013-04-499491; PMID: 23861251
- Nurden AT, Nurden P, Bermejo E, Combrié R, McVicar DW, Washington AV. Phenotypic heterogeneity in the Gray platelet syndrome extends to the expression of TREM family member, TLT-1. Thromb Haemost 2008; 100:45 - 51; PMID: 18612537
- Morowski M, Vögtle T, Kraft P, Kleinschnitz C, Stoll G, Nieswandt B. Only severe thrombocytopenia results in bleeding and defective thrombus formation in mice. Blood 2013; 121:4938 - 47; http://dx.doi.org/10.1182/blood-2012-10-461459; PMID: 23584880
- Bottega R, Pecci A, De Candia E, Pujol-Moix N, Heller PG, Noris P, De Rocco D, Podda GM, Glembotsky AC, Cattaneo M, et al. Correlation between platelet phenotype and NBEAL2 genotype in patients with congenital thrombocytopenia and α-granule deficiency. Haematologica 2013; 98:868 - 74; http://dx.doi.org/10.3324/haematol.2012.075861; PMID: 23100277
- Bertozzi CC, Hess PR, Kahn ML. Platelets: covert regulators of lymphatic development. Arterioscler Thromb Vasc Biol 2010; 30:2368 - 71; http://dx.doi.org/10.1161/ATVBAHA.110.217281; PMID: 21071706
- Bergmeier W, Stefanini L. Platelet ITAM signaling. Curr Opin Hematol 2013; 20:445 - 50; http://dx.doi.org/10.1097/MOH.0b013e3283642267; PMID: 23921514
- Goerge T, Ho-Tin-Noe B, Carbo C, Benarafa C, Remold-O’Donnell E, Zhao B-Q, Cifuni SM, Wagner DD. Inflammation induces hemorrhage in thrombocytopenia. Blood 2008; 111:4958 - 64; http://dx.doi.org/10.1182/blood-2007-11-123620; PMID: 18256319
- Burgess A, Mornon JP, de Saint-Basile G, Callebaut I. A concanavalin A-like lectin domain in the CHS1/LYST protein, shared by members of the BEACH family. Bioinformatics 2009; 25:1219 - 22; http://dx.doi.org/10.1093/bioinformatics/btp151; PMID: 19289442
- Cullinane AR, Schäffer AA, Huizing M. The BEACH is hot: a LYST of emerging roles for BEACH-domain containing proteins in human disease. Traffic 2013; 14:749 - 66; http://dx.doi.org/10.1111/tra.12069; PMID: 23521701
- Urban D, Li L, Christensen H, Pluthero FG, Chen SZ, Puhacz M, Garg PM, Lanka KK, Cummings JJ, Kramer H, et al. The VPS33B-binding protein VPS16B is required in megakaryocyte and platelet α-granule biogenesis. Blood 2012; 120:5032 - 40; http://dx.doi.org/10.1182/blood-2012-05-431205; PMID: 23002115
- Nurden P, Gobbi G, Nurden A, Enouf J, Youlyouz-Marfak I, Carubbi C, La Marca S, Punzo M, Baronciani L, De Marco L, et al. Abnormal VWF modifies megakaryocytopoiesis: studies of platelets and megakaryocyte cultures from patients with von Willebrand disease type 2B. Blood 2010; 115:2649 - 56; http://dx.doi.org/10.1182/blood-2009-07-231886; PMID: 20118404