Abstract
RNA viruses exhibit small-sized genomes encoding few proteins, but still establish complex networks of protein-protein and RNA-protein interactions within a cell to achieve efficient replication and spreading. Deciphering these interactions is essential to reach a comprehensive understanding of the viral infection process. To study RNA-protein complexes directly in infected cells, we developed a new approach based on recombinant viruses expressing tagged viral proteins that were purified together with their specific RNA partners. High-throughput sequencing was then used to identify these RNA molecules. As a proof of principle, this method was applied to measles virus nucleoprotein (MV-N). It revealed that in addition to full-length genomes, MV-N specifically interacted with a unique population of 5′ copy-back defective interfering RNA genomes that we characterized. Such RNA molecules were able to induce strong activation of interferon-stimulated response element promoter preferentially via the cytoplasmic pattern recognition receptor RIG-I protein, demonstrating their biological functionality. Thus, this method provides a new platform to explore biologically active RNA-protein networks that viruses establish within infected cells.
Introduction
Viruses have evolved specific proteins to bind their genomic material. Nucleocapsid proteins typically enwrap viral genomes for protection and transport but also promote viral transcription and replication steps through interactions with viral polymerase components. Numerous studies have shown that viral proteins also bind viral sub-genomic as well as cellular nucleic acids to achieve efficient replication.Citation1-Citation4 However, a technological framework to investigate such interactions in a comprehensive manner within infected cells is still missing. High-throughput sequencing allows the characterization of nucleic acid compartments within protein-DNA or protein-RNA complexes. Chromatin immunoprecipitation followed by sequencing (ChIP-seq) can provide genome-scale information on DNA-protein interactions, whereas RNA-protein interactions have been investigated using RNAs isolated by the crosslinking and immunoprecipitation (HITS-CLIP) approach.Citation5 Recently, immunoprecipitation and next-generation sequencing have also been applied to study in an infectious context viral RNA interactions with retinoic acid inducible gene I (RIG-I), a cellular sensor that triggers immune response against viruses.Citation6 To our knowledge, to date no approach has been applied to perform high-throughput characterization of RNA partners of viral proteins during infection.
Measles virus (MV), a member of the order Mononegavirales possesses a negative, single-stranded RNA genome. The nucleoprotein of MV (MV-N) efficiently encapsidates the virus genome into a helical viral nucleocapsid (viral ribonucleoprotein, RNP), and this protein-RNA complex is used as template by the viral RNA-dependent RNA polymerase for both transcription and replication.Citation7 MV replication is coupled to encapsidation so that full-length antigenomes and genomes are found only inside assembled nucleocapsids.Citation8 Specific encapsidation signals, which are recognized by MV-N, lie within the 5′ Leader (Le) and Trailer (Tr) extremities of the antigenome and genome RNA strands.Citation7 In addition to infectious particles containing a full-length viral genome, MV produces defective interfering (DI) particles containing only a portion of the virus genetic information, usually referred as DI genomes (or DI-RNAs).Citation9,Citation10 DI particles cannot replicate in host cells unless they co-infect target cells with a competent helper virus that supplies the missing gene products. Finally, in the absence of viral RNA, nucleoproteins of Mononegavirales are able to self-assemble onto cellular RNA to form nucleocapsid-like particles and this self-assembly is believed to be sequence independent.
We have recently shown that virus reverse genetics can be efficiently combined with mass spectrometry-based analysis to characterize protein partners of a viral protein in infected cells.Citation11 We now report a strategy to characterize, in an infectious context, the nucleic acids bound to a viral protein. Due to its RNA-binding activity, as proof of concept, the MV-N protein was chosen and we used a recombinant MV strain expressing a tagged MV-N protein.Citation11 This recombinant virus allowed MV-N purification together with its partners directly from infected cells using a one-step purification approach. In the present study, the pool of RNA molecules that co-purified with nucleocapsids were extracted, next-generation sequencing was performed and MV-N RNA-interacting partners were characterized. To our knowledge, this is the first report that provides an integrated strategy to identify interactions between viral proteins and RNA molecules inside infected cells. In the future, the same method could be applied to any protein of interest from any virus for which reverse genetics is available.
Results
Recombinant MV strain encoding an additional C-terminally tagged MV-N protein
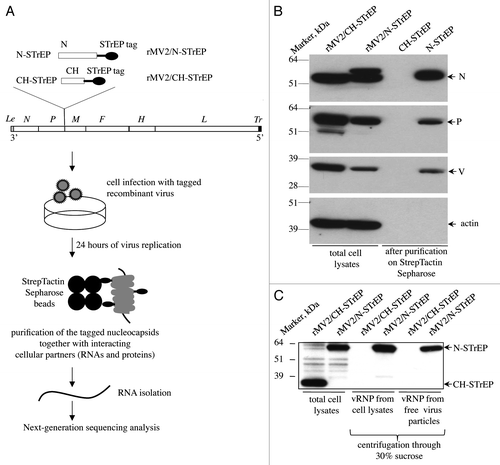
To analyze the RNA compartment of MV nucleocapsids, a reverse genetics approach was used and a recombinant MV strain expressing an additional tagged MV-N protein was engineered. Since a previous report has demonstrated that the known MV-N functions were not affected by introduction of the a C-terminal tag whereas a N-terminal tag was deleterious,Citation12 a sequence encoding the MV-N protein fused to a C-terminal One-STrEP tag (see Materials and Methods) was introduced in an additional transcription unit (ATU) within the MV genome (). We previously used this recombinant virus (rMV2/N-STrEP) to study cellular proteins that interact with MV nucleocapsids in virus-infected cells.Citation11 In addition, as negative control, a recombinant virus expressing the Cherry (CH) protein was used instead of MV-N (rMV2/CH-STrEP, ). The growth kinetics and titers of rMV2/N-STrEP and rMV2/CH-STrEP were similar to unmodified MV Schwarz vaccine strain.Citation11 In this system and as expected, MV-N-STrEP co-purified with the MV proteins P and V, but not with cellular actin that served as negative purification control (). As additional control, we also showed that MV-N, P and V did not co-purify with CH-STrEP from cells infected with rMV2/CH-STrEP. This confirms that One-STrEP-tagged MV-N specifically interacts with known interactors of MV-N. Furthermore, we established that One-STrEP-tagged MV-N proteins are efficiently incorporated into viral RNPs, both within infected cells and inside cell-free viral particles, by analyzing on western blots the composition of viral RNPs condensed by ultracentrifugation through 30% sucrose (). These results strongly suggest that by purifying One-STrEP-tagged MV-N, it is extracted in a form of viral ribonucleocapsid. In contrast, the negative control One-STrEP-tagged CH protein was efficiently expressed by rMV2/CH-STrEP but failed to incorporate into viral ribonucleocapsids ().
Figure 1. Validation of recombinant MV expressing 1-STrEP-tagged MV-N and the protocol used to purify specific RNA partners of MV-N. (A) Schematic representation of rMV2/N-STrEP and rMV2/CH-STrEP recombinant viruses and summary of the protocol used to purify MV-N and associated RNA molecules from infected cells. The MV genome is displayed with the six genes indicated by italicized capital letters. The position of One-STrEP-tagged MV-N or CH protein insertion is shown. The black oval represents the One-STrEP-tag sequence. MV leader (Le) and trailer (Tr) regions are shown as gray and dark rectangles, respectively. (B) Analysis of MV-N viral protein-partners within an infected cell. Actin and CH proteins served as negative controls to see the specificity of the MV-N complexes after modified tandem affinity purification.Citation11 WB analysis using anti-N, anti-P, anti-V and anti-actin antibodies. (C) Ultracentrifugation through 30% sucrose followed by western blot analysis shows efficient incorporation of the N-STrEP protein within the viral nucleocapsid. Viruses used for infection are indicated above each lane. One-STrEP-tagged proteins were detected using Streptavidin-HRP.
Isolation of MV-N and associated RNA molecules from virus-infected cells
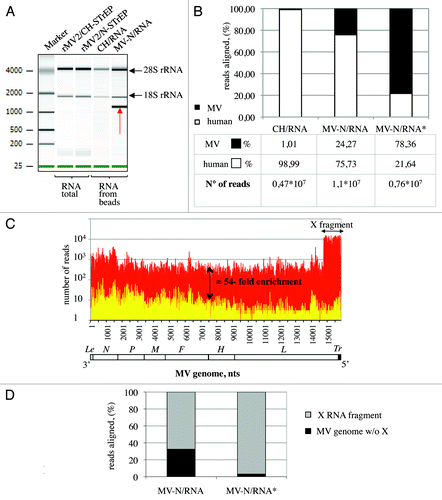
The entire purification protocol is presented as a flowchart in . To co-purify MV-N and associated RNA molecules, HEK-293T cells were infected with rMV2/N-STrEP virus. After 24 h of culture, the cells were harvested and One-STrEP-tagged MV-N together with associated partners was purified from total cell lysates by affinity chromatography using StrepTactin-conjugated Sepharose beads. The RNAs interacting with MV-N were then extracted from the beads. To distinguish between MV-N-associated RNA molecules and non-specific binding to the beads, co-affinity purification experiments were performed in parallel with rMV2/CH-STrEP virus. RNA molecules that co-purified with MV-N or CH-tagged proteins, designated MV-N/RNA and CH/RNA samples, were compared with total RNAs isolated from either rMV2/N-STrEP or rMV2/CH-STrEP-infected cells by horizontal agarose gel electrophoresis (not shown) or capillary electrophoresis using the Agilent technology (). The total RNA profiles were similar. After co-affinity purification, both MV-N/RNA and CH/RNA samples revealed a substantial fraction of rRNA. This can be explained by non-specific binding of abundant cellular RNAs to the StrepTactin Sepharose beads used for purification. However, the profile of RNA molecules that co-purified with One-STrEP-tagged MV-N presented a prominent and specific band slightly above 1,000 nucleotides (), indicating specific binding of RNA species to MV-N that called for in-depth characterization.
Figure 2. Analysis of purified MV-N RNA partners. (A) Agilent eukaryotic total RNA assay of MV-N/RNA samples reveals a distinct band slightly above 1,000 nucleotides. Total RNA purified from cells infected with rMV2/CH-STrEP or rMV2/N-STrEP viruses or RNA samples after purification on StrepTactin Sepharose beads. (B) Comparison of number of sequences that mapped to MV or human genomes for CH/RNA, and MV-N/RNA samples. MV-N/RNA*sample illustrates the data obtained when the X RNA fragment was excised from the polyacrylamide gel before deep sequencing analysis. (C) High-throughput sequencing analysis of MV-N-associated RNA from MV-infected cells. MV-N/RNA and CH/RNA from MV-infected cells (red and yellow, respectively) were subjected to Illumina high-throughput sequencing. Sequencing reads were mapped to the virus genome and only the first nucleotide was retained. The Y-axis shows in logarithmic scale the number of reads that begin at a particular position. (D) Comparison of the number of sequences that map to the X RNA fragment of the MV genome and to the rest of the MV genome for the MV-N/RNA and MV-N/RNA* samples.
Characterization of RNA species bound to MV nucleocapsids by next-generation sequencing
To identify the nature of RNA molecules bound to MV-N in an unbiased manner, high-throughput sequencing analysis was performed using the Illumina technology. Sequences were mapped to human and MV genomes, and the fraction of reads aligning to each genome was determined. As shown in , RNA molecules that co-purified with MV-N showed a strong enrichment in viral sequences when compared with the CH/RNA sample. This can be explained by the preferential association of MV genomes and anti-genomes to MV-N within viral RNP complexes and suggests that specific MV-N-associated RNAs are most likely of viral origin. We then normalized libraries using the corresponding TMM (Trimmed Mean of M values) scaling normalization factors proposed by Robinson and OshlackCitation13 and used them to normalize read counts as explained in Materials and Methods. To search for cellular RNA partners of MV-N, the distributions of normalized read counts obtained for the two RNA samples (MV-N/RNA, CH/RNA) were aligned on the human genome. In addition, the Fisher exact test followed by a Benjamini-Hochberg p value adjustment was used to detect positions on the human genome that had significantly different read counts between the two samples. However, except for several non-coding RNAs, such as 5S rRNA, no prominent cellular RNA partner for MV-N was detected (data not shown). Thus, specific viral RNA partners were sought.
The distribution of normalized read counts matching the MV sequence was represented along the viral genome with the X axis corresponding to all possible positions on the MV genome, and the Y axis showing the number of reads that begin at that position (). When MV-N/RNA and CH/RNA samples were mapped to the MV genome, the MV-N/RNA sample showed an average 54-fold enrichment for MV reads all along the MV genome. This demonstrated the preferential association of the MV genome and anti-genome to MV-N within viral nucleocapsids. Interestingly, a 1,114 nucleotide-long sequence that mapped to the 5′ end of the MV genome (assigned as X RNA fragment) was 15 times more abundant in MV-N/RNA samples compared with the rest of the viral genome (; Table S1). This RNA fragment was not detected in CH/RNA samples and probably corresponds to the RNA fragment visualized using Agilent analysis (). To validate this hypothesis, molecules in the MV-N/RNA samples were separated by PAGE, and the band of about 1,100 nucleotides was excised and characterized by high-throughput sequencing. Most sequencing reads obtained from this gel-purified RNA aligned to the MV genome (; MV-N/RNA*), and a vast majority corresponded to the last 1,114 nucleotides of the MV genome, thus matching the X RNA fragment (). The presence of this RNA species in MV-N/RNA samples could be explained by (1) a preferential interaction with MV-N over the full-length MV genomes, (2) an over-production of this RNA species by the rMV2/N-STrEP recombinant virus or (3) both mechanisms. Interestingly, previous studies have mapped 5′ copy-back defective interfering RNA genomes (DI-RNA) to the 5′ end of MV genomes.Citation9,Citation10 This suggested that the X RNA fragment could correspond to DI-RNA molecules. To address this question, the exact sequence of this X RNA fragment was determined.
Characterization of the 5′ copy-back RNA molecule and validation of the specificity of its interaction with the MV-N protein
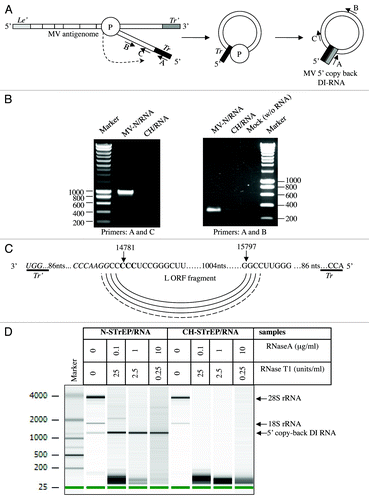
5′ copy-back DI-RNAs can be produced by MV and other paramyxoviruses and, in particular, vaccine strains, during their replication cycle.Citation9 Such DI-RNAs consist of the genomic “trailer” (Tr) sequence, a fragment of the L gene and the reverse complement of the Tr sequence (Tr’) ().Citation14 To determine if the X RNA species over-represented in purified nucleocapsid reads corresponded to DI-RNA molecules, samples were reverse-transcribed and amplified by PCR (RT-PCR) with DI-RNA-specific primers (see Materials and Methods and ref. Citation9). Amplicons were obtained when RT-PCR was performed on MV-N/RNA samples, but not on CH/RNA samples (). Furthermore, sequencing analysis revealed that the RT-PCR amplicon contains all the DI-RNA features described above, including the exact complementary extremities of 98 nucleotides able to hybridize to form a stem-loop structure (). Most importantly, this allowed us to determine that premature termination of negative strand synthesis occurs exactly at position 14,781 of the MV genome, after which the viral polymerase starts synthesis of the antigenome sequence at position 15,797 of the MV genome resulting in a single population of RNA molecules exactly 1,212 nucleotide long (). In addition, the 1,212 nucleotide-long 5′ copy-back DI-RNA efficiently resisted RNase digestion under conditions that degraded cellular RNP complexes such as ribosomes ().
Figure 3. Characterization of the 5′ copy-back RNA molecule interacting with MV-N. (A) Model for the origin of the 1,212 nucleotide-long DI-RNA of MV. Following premature termination of (-) strand synthesis, the polymerase, with the nascent strand attached, resumes synthesis at a specific internal site and forms the complementary “stem” structure. Anti-leader (Le’) and anti-trailer (Tr’) sequences are shown as light gray and dark gray rectangles, respectively, and trail sequence (Tr), as a black rectangle. “P” stands for the viral polymerase complex. Positions and directions of A, B and C primers are shown. (B) RT-PCR amplification of the 5′ copy-back DI-RNA or the MV genome fragment from the MV-N/RNA complex and CH/RNA control. RT-PCRs were performed using DI-RNA-specific primers (A and C) or standard genome-specific primers (A and B) (see Materials and Methods and ref. Citation9). (C) Exact sequence of the 5′ copy-back 1,212 nucleotide-long DI genome of MV. Nucleotides at the position where viral polymerase resumes synthesis to transcribe the complementary “stem” structure are indicated and shown in bold. (D) Agilent eukaryotic total RNA analysis of MV-N/RNA samples shows protection by MV-N protein of a bulk of the 5′ copy-back DI-RNA from digestion by RNase A and RNase T1. MV-N and CH RNA samples after purification on StrepTactin Sepharose beads were incubated with serial dilutions of RNase A/T1 mix before RNA extraction. The table above the figure shows RNase A and T1 concentrations in each sample.
Viruses with both RNA and DNA genomes are capable of spontaneously generating DI genomes during their replication cycle. They are believed to accumulate by serial passage of the virus at high multiplicities of infection (MOI), culture on specific host cells, laboratory adaptation of wild-type viruses to cell culture or insertion of specific mutations within the virus genome (for a review, see refs. Citation14 and Citation15). However, the precise conditions that determine the production of DI-RNA genomes are poorly characterized. We first determined by one-step reverse transcription-quantitative PCR (RT-qPCR) whether the production of the 1,212 nucleotide-long MV DI-RNA was cell-dependent. Using four different cell lines (Vero, HEK293T, A549, Hela), we observed that this was not the case (). Furthermore, we compared DI-RNA levels produced by rMV2/N-STrEP, or a low-passage Schwarz vaccine strain of MV, or the Schwarz vaccine strain grown for five consecutive passages at a MOI of 1. Surprisingly, insertion of the second copy of MV-N ORF within the MV genome as performed in the rMV2/N-STrEP strongly enhanced production of DI-RNAs compared with the original Schwarz virus. Moreover, as few as five passages were sufficient to increase DI-RNA production by Schwarz MV (). Interestingly, almost no difference in production of the full-length genome was detected ().
Figure 4. In spite of strong enhancement of the MV DI-RNA production due to the insertion of the second copy of the N protein within the MV genome, the 5′ copy-back DI-RNA specifically interacts with the MV nucleocapsid. (A) Efficient production of the 1,212 nucleotide-long 5′ copy-back MV DI-RNA by rMV2/N-STrEP in four different cell lines. Total RNA (20 ng) was analyzed by RT-qPCR. Absolute quantification of DI-RNA was performed using serial dilutions of in vitro transcribed MV DI-RNA. Samples were analyzed in triplicates, with standard deviation represented on the figure, and two biological replicates were performed each time. (B) Production of the 1,212 nucleotide DI genome and the full-length genome by recombinant MV expressing the second copy of MV-N. 293T cells were infected with either rMV2/N-STrEP, or a low-passage Schwarz vaccine strain of MV, or a Schwarz vaccine strain grown for five consecutive passages at a MOI of 1. Total RNA was purified and 1 μg of each RNA sample was analyzed by RT-qPCR against either the 1,212 nucleotide-long DI-RNA or the full-length MV genome. Absolute quantification was performed using serial dilutions of the in vitro transcribed MV DI-RNA or the MV genome RNA fragment. Samples were analyzed in triplicates, with standard deviation represented on the figure, and two biological replicates were performed each time. (C) Affinity chromatography of 1-STrEP tagged viral proteins form infected cells is an efficient and specific approach to purify RNA partners of viral proteins. Results of RT-qPCR against the 1,212 nucleotide-long DI-RNA on 10 ng of RNA purified complexed with the MV-N protein were compared with data obtained for 10 ng of total RNA (before purification). Absolute quantification was performed as described in (A). As negative control one-step RT-qPCR against the β-actin mRNA was performed with absolute quantification using serial dilutions of in vitro transcribed RNA fragments encompassing the β-actin sequence. Samples were analyzed in triplicates and two biological replicates were performed.
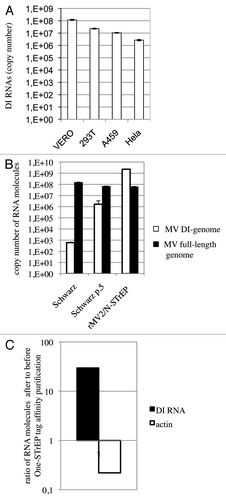
Since the rMV2/N-STrEP virus massively expressed DI-RNAs, we wished to determine if these RNA molecules were not only overproduced but were, as is the MV genome, specifically bound to MV-N over other cellular RNA molecules. Using one-step RT-qPCR absolute quantification of the 5′ copy-back DI-RNA was performed before and after One-STrEP tag affinity purification. This experiment showed that DI-RNA was enriched about 30 times after purification compared with total RNA sample (). As negative control, the highly abundant β-actin mRNA was used that showed a strong depletion after 1-STrEP tag affinity purification (). Thus, despite the overproduction of DI-RNA species by rMV2/N-STrEP, this result confirmed specific binding of DI-RNA to MV-N as compared with cellular RNA molecules.
The 1,212 nucleotide-long 5′ copy-back MV DI-RNA triggers efficient interferon induction via RIG-I
5′ copy-back DI-RNA molecules from negative-strand RNA viruses are known for their ability to induce type-I interferon (IFN) signaling as demonstrated by direct and indirect approaches.Citation6,Citation9,Citation16-Citation18 Type I IFN is induced in all cell types by receptors that monitor the cytosol for the presence of nucleic acids indicative of virus presence. Such sensors include RIG-I-like receptors that recognize RNA and are themselves IFN-inducible (for a review, see ref. Citation19). Two members of this family, RIG-I and MDA5 (melanoma differentiation-associated gene-5), possess two N-terminal caspase activation and recruitment domains that allow interaction with the mitochondrial antiviral-signaling protein (MAVS). MAVS triggers the activation of several transcription factors, including NFκB, IRF-3 and IRF-7, which, in turn, induces transcription of type-I IFN and other innate response genes.
To demonstrate the biological functionality of RNA molecules that co-purified with One-STrEP-tagged MV-N, their capacity to induce an interferon-mediated antiviral response was evaluated. MV-N/RNA and CH/RNA samples were transfected into the STING-37 reporter cell line. This cell line stably expresses a firefly luciferase gene under the control of a promoter sequence containing five IFN-stimulated response elements (M. Lucas-Hourani to be published elsewhere). Transfection of MV-N-associated RNA into reporter cells resulted in a 16-fold increase in reporter activity compared with the CH control (). In contrast, the immunostimulatory activity of total RNA samples purified from cells infected with rMV2/N-STrEP or rMV2/CH-STrEP failed to show any difference (data not shown). This indicates that the RNA molecules specifically purified in association with the MV-N protein and enriched in 5′ copy-back MV DI-RNA had a significant immunostimulatory activity, in agreement with previous results.Citation6,Citation9,Citation16,Citation17
Figure 5. The 1,212 nucleotide-long 5′ copy-back MV DI-RNA triggers efficient IFN type I production via RIG-I. (A) Immunostimulatory activity of RNA from MV-N upon transfection into STING-37 cells. MV-N affinity chromatography-purified RNAs were compared with RNAs purified from negative control (CH)-tagged protein. Samples were analyzed in triplicates with standard deviation represented on the figure, and two biological replicates were performed. (B) The 1,212 nucleotide-long DI genome of MV induces RIG-I mediated type-I IFN signaling. STING-37 reporter cells were transfected with siRNA targeting RIG-I, MDA-5, MAVS or TPR (negative control) or mock transfected. Second transfection of the cells was performed 48 h later with either 1,212 nucleotide-long MV DI-RNA, poly(IC) or 5′PPP-bearing short RNA. Luciferase induction fold is expressed as the ratio of luciferase measured within stimulated by second RNA transfection cells to the ratio of luciferase in cells left without the second transfection. Samples were analyzed in triplicates and two biological replicates were performed. The SEMs are shown in the figure.
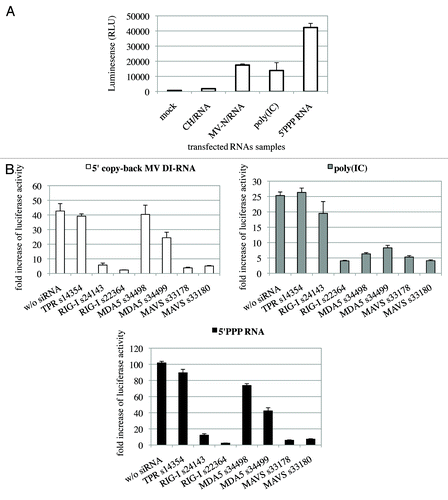
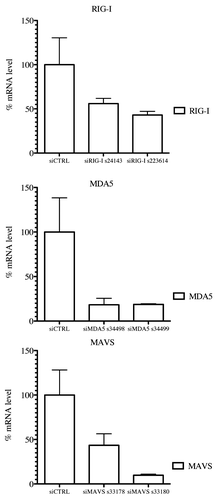
Earlier, Shingai et al. reported that DI-RNA genomes produced by vaccine strains of MV induce IFN-β production as a consequence of DI-RNA detection by cellular cytoplasmic sensors such as MDA5 and RIG-I.Citation9 We tested whether the 1,212 nucleotide-long 5′ copy-back MV DI-RNA described here exerts its immunostimulatory activity via RIG-I and/or MDA5 cytoplasmic sensors. DNA sequences encoding the full-length 5′ copy-back DI-RNA molecule were cloned downstream of a T7 promoter. The RNA fragment was transcribed in vitro and co-transfected into reporter STING-37 cells, in the presence or absence of siRNA targeting RIG-I, MDA5 or MAVS ( and ). To determine the induction of the IFN-α/β antiviral response by DI-RNA, the ratio of luciferase activity in DI-RNA-transfected cells over the background of luciferase activity in un-transfected cells was determined. DI-RNA molecules efficiently induced type-I IFN response as assessed by luciferase expression, and RIG-I was confirmed to be the receptor involved. Thus, the combination of affinity chromatography and next-generation sequencing allowed us to identify the specific MV-N-interacting RNA molecule that interplays with the host cellular innate immunity.
Figure 6. RIG-I, MDA5 and MAVS mRNA levels in knockdown STING-37 cells. Total RNA was extracted from cells previously transfected with siRIG-I, siMDA5 or siMAVS STING-37 cell line, and the levels of RIG-I, MDA5 and MAVS mRNA were quantified by one-step RT-qPCR. The data were normalized to the GAPDH mRNA levels and expressed as relative levels of mRNA compared with cells without siRNA transfection (siCTRL). The mean value in STING-37 cells mock-transfected with siRNA was set at 100% of gene expression.
Discussion
High-throughput protocols such as yeast-two-hybrid or MS-based protein-complex analysis have been extensively used to generate virus-host protein-protein interaction data.Citation11,Citation20-Citation22 Recently, we have used reverse genetics to engineer the MV genome and rescue recombinant viruses that encode either tagged MV-V or MV-N proteins. These viruses have allowed virus-host protein complex analysis directly from infected cells by combining modified tandem affinity chromatography and mass spectrometry analyses. Using this approach, a prosperous list of 245 cellular proteins was established that interact either directly or indirectly with MV-V. In contrast, only eight unique cellular partners were identified that interact with MV-N,Citation11 suggesting that the MV nucleocapsid stays rather inert with respect to interaction with host proteins. However, the MV-N protein is also known for its RNA-binding activity (for a review, see ref. Citation7). In the present study, we aimed to map virus-host RNA-protein interactions, and developed a new method based on recombinant viruses expressing tagged viral proteins, affinity chromatography purification and next-generation sequencing (). This approach made it possible to capture specific RNA-binding partners of MV nucleocapsids and to identify them. Except for several abundant cellular noncoding RNAs, no prominent cellular RNA partner for MV-N was detected and we encountered difficulties in validating these interactions using a conventional RT-qPCR approach. Thus, we searched for specific viral RNA partners and found that in addition to the full-length MV genome, the MV-N protein specifically interacted with the 1,212 nucleotide-long RNA molecule corresponding to 5′ copy-back DI genomes (). We confirmed that this RNA molecule was specifically enriched within the MV-N/RNA after One-STrEP tag affinity chromatography purification as compared with total RNA before purification (). Finally, its functional role in activating type-I IFN response was confirmed and showed that this activation occurred preferentially via the cytoplasmic pattern recognition receptor RIG-I ().
New DNA sequencing technologies, such as next-generation sequencing or high-throughput sequencing, can sequence up to one billion bases in a single day at low cost with no need to design specific primers (for a review, see refs. Citation5 and Citation23). Next-generation sequencing is a powerful tool for virology, from the investigation of viral diversity to the discovery of new viral species without any advance genetic information. This approach was recently employed to compare the RNA encapsidated by authentic Flock House virions to virus-like particles and demonstrated that the genetic content of non-enveloped RNA viruses was variable, not just due to genome mutations, but also to the diversity of the host RNA transcripts that were packed inside viral particles.Citation24 High-throughput sequencing of RNAs isolated by immunoprecipitation (preceded or not by UV crosslinking) is emerging as an important method to understand RNA-protein interactions.Citation6,Citation25 However, immunoprecipitation of RNA-protein complexes relies on the availability of high-affinity antibodies directed to epitopes that remain accessible within the complex. As an alternative of immunoprecipitation, in the present study we suggest using recombinant viruses expressing tagged viral proteins to perform affinity chromatography purification of RNP complexes from infected cells. Additionally, to distinguish between the viral protein-associated RNA molecules and non-specific binding to the beads, a recombinant MV strain was used expressing a One-STrEP-tagged CH protein. This control was efficient to distinguish between specific MV nucleocapsid interactions and non-specific binding to beads with average ratio of MV-N/RNA reads to CH/RNA reads of about 50 outside the DI-RNA region (; Table S1). Thus, applying affinity chromatography purification of tagged viral proteins encoded by the viral genome allowed efficient purification of specific RNA partners for viral proteins.
Though being an attractive method to study RNA-protein interactions during viral infection, manipulation of viral genomes by reverse genetics could lead to some differences in viral replication. Previous studies have shown that fusion of GFP C-terminally to MV-N and introduction of the fused MV-N-GFP in an ATU only slightly delayed growth of the recombinant virus and lacked a negative effect on MV-N-GFP incorporation into viral particles. At the same time, the recombinant MV expressing MV-N-GFP instead of N has failed to be rescued showing that both the N- and C-terminal parts of the nucleoprotein should be intact to provide efficient viral replication.Citation12 Based on this study, we have previously introduced C-terminally tagged MV-N in an ATU within the MV genome and demonstrated that the recombinant virus growth kinetics was similar to that of unmodified MV Schwarz.Citation11 In the present study, the MV-N-tagged protein was efficiently incorporated into viral RNP (). On the other hand, insertion of tagged nucleoprotein within the ATU strongly enhanced the production of the 1,212 nucleotide-long DI-RNA (). This suggests that an additional copy of MV-N somehow perturbed the MV replication complex leading to massive production of the DI genomes. Nevertheless, and despite such an overproduction of DI-RNA species, the specific binding of these molecules to MV-N was confirmed ().
Previous studies suggested that the likeliest mechanism for the generation of DI genomes was an error of the viral polymerase, “skipping” from one template to another or from one part of a template to another, and then resuming elongation on the 3′ end of the nascent strand which had been synthesized prior to the “skip.”Citation14,Citation15 Using next-generation sequencing of purified complexes, we demonstrated for the first time that the MV DI-RNA encapsidated by MV-N is a single population of exactly 1,212 nucleotide-long RNA molecules ( and ; Table S1). Thus, the MV RNA-dependent RNA-polymerase starts the production of the 5′ copy-back DI-RNA when it encounters the specific sequence on the MV genome (). Hence, in addition to the known sequence motifs recognized by the MV polymerase complex to perform replication, transcription, polyadenylation and non-template insertion of extra G residues to generate V and W transcripts,Citation26 yet unknown signals on the MV genome lead to the production of the 5′ copy-back DI-RNA. Additionally, for Paramyxovirinae (a subfamily of the Paramyxoviridae), each nucleoprotein wraps exactly six nucleotides that imposes the so-called “rule of six” to these viruses, i.e., their genome must be of polyhexameric length to efficiently replicate.Citation27 The MV 5′ copy-back DI-RNA identified here is a multiple of six nucleotides (6 × 202 = 1,212) as two other well-characterized examples of 5′ copy-back DI-RNA of 546 nucleotide-long and 1,410 nucleotide-long RNAs observed for Sendai virus, another member of the Paramyxovirinae.Citation6,Citation28 This suggests that DI genomes of Paramyxovirinae, as also full-length genomes, follow the rule of six to achieve efficient encapsidation and replication. Indeed, we performed an RNase protection assay and observed that the 5′ copy-back DI genome was efficiently encapsidated by MV-N (), similarly to the full-length viral genome.Citation29,Citation30 Additional studies should be performed to uncover the mechanism of DI-RNA production. Moreover, recombinant viruses efficiently expressing DI genomes might be of great practical importance for vaccinology due to their ability to stimulate the innate immunity response. Indeed, the 1,212 nucleotide-long DI-RNA efficiently induced a type-I IFN response preferentially via the cytoplasmic pattern recognition receptor RIG-I protein ().
Taken together, our data demonstrate the advantage of combining reverse genetics with affinity chromatography purification of tagged viral proteins in the form of RNP complexes, RNA extraction and next-generation sequencing of viral RNA partners. Our method is unique because interactions between viral proteins and RNA molecules can be investigated inside infected cells and throughout the virus replication cycle. This is different from the study by Routh et al. in which only assembled virions or virus-like particles were purified and analyzed for their association with RNA molecules.Citation24 In the future, a similar strategy could be applied to other viruses for which tagged viral proteins can be introduced into the viral genome or expressed within a cell via transfection in the presence of an appropriate viral infection. Finally, as an alternative to immunoprecipitation followed by next-generation sequencing, affinity purification of RNA-protein complexes could easily be applied to study RNA partners of cellular RNA-binding proteins. Thus, our method provides a relevant strategy to perform high-throughput identification of biologically active RNA-protein interactions inside a cell.
Materials and Methods
Cells and recombinant viruses
HEK-293T (human embryonic kidney) and Vero cells (African green monkey kidney cells) were maintained in DMEM-Glutamax (Gibco-BRL) supplemented with 10% heat-inactivated fetal calf serum (Invitrogen). The STING-37 cell line corresponding to HEK-293 cells stably transfected with an ISRE-luciferase reporter gene will be described in detail elsewhere (M. Lucas-Hourani et al., manuscript in preparation). Briefly, the ISRE-luciferase reporter gene was amplified by PCR from the pISRE-luciferase reporter plasmid (#219089, Stratagene), and inserted into a plasmid carrying a G418-resistance selection marker. This plasmid was transfected into HEK-293 cells (ATCC) and 2 d later, the culture medium was supplemented with G418 at 400 μg/ml. Transfected cells were amplified and subsequently cloned by serial limit dilution. A total of 44 clones were screened for luciferase expression, and the STING-37 clone was selected for its optimal signal to background ratio whether stimulated or not with recombinant IFN-β. The MV Schwarz vaccine strain and two recombinant MVs expressing an additional N or CH protein with a C-terminal STrEP-tag (ENLYFQSEASWSHPQFEKGGGSGGGSGGGSWSHPQFEKGA, the Tobacco Etch virus protease linker and STrEP-tag protein sequence are shown in bold) inserted in an additional transcription unit between the P and M genes and designated rMV2/N-STrEP and rMV2/CH-STrEP () were described elsewhere.Citation11,Citation31
Identification of MV-N virus protein partners
Purification of MV-N together with its protein partners was performed as described.Citation11 Protein extracts before and after affinity chromatography purification were resolved by SDS-PAGE (PAGE) on 4–12% NuPAGE Bis-Tris gels (Invitrogen) with MOPS running buffer (Invitrogen) and transferred to cellulose membranes with the Criterion Blotter system (BioRad). Membranes were blotted with either a mouse anti-N mAb (clone 25) antibody (kindly provided by Pr. Chantal Rabourdin-CombeCitation32), a rabbit anti-V antibodies (kindly provided by Dr. Kaoru TakeuchiCitation33), Measles Phospho-protein Antibody (#9H4, Novus) antibody or anti-β-actin antibody (#A5441, Sigma). Peroxidase activity was visualized with an ECL Plus Western Blotting Detection System (#RPN2132, GEHealthcare).
Purification of recombinant viral RNPs from infected cells and free viral particles
Recombinant viral RNPs were purified from confluent monolayers of HEK-293T cells infected with rMV2/N-STrEP or rMV2/CH-STrEP at a MOI of 0.1. Infected cells (3 × 107) were harvested by centrifugation at 200 g and 8 ml of each culture medium fractions were collected for analysis of cell-free viral particles. Collected cells washed three times with cold PBS and cell pellets were resuspended in hypotonic buffer (10 mM TRIS-HCl pH 7.5, 10 mM NaCl) at 1 × 107 cells/ml in the presence of a protease inhibitor cocktail (#11873580001, Roche). 1% final of Igepal (#I8896, SigmaAldrich) was added to both cell and culture medium fractions and the fractions were left on ice for 30 min for lysis. Cell and culture medium fractions were then centrifuged at 1,000 g at 4°C for 5 min, and the supernatants were collected and adjusted to contain 10 mM EDTA. These fractions were also clarified by centrifugation at 10,000 g at 4°C for 10 min. Further clarified culture media and cytoplasmic extracts were ultracentrifuged through 3 ml of 30% (wt/vol) saccharose each containing 25 mM TRIS-HCl pH 7.5, 50 mM NaCl and 2 mM EDTA. The ultracentrifugation was performed at 4°C for 2 h at 36,000 rpm in an SW 41 swinging bucket rotor. The supernatant was removed and the transparent viral RNP pellet was solubilized in 0.1 ml of western blot loading buffer. Viral RNP proteins were resolved by SDS-PAGE on 4–12% NuPAGE Bis-Tris gel (Bio-Rad) with MOPS running buffer and transferred to a cellulose membrane with the Criterion blotter system (BioRad). To detect One-STrEP-tag the membrane was blotted with Streptavidin-HRP (#19534-050, Invitrogen).
One-STrEP tag purification of MV-N/RNA and CH/RNA complexes and subsequent RNA extraction
HEK-293T cells (3.6 × 108) were infected with rMV2/N-STrEP or rMV2/CH-STrEP at a MOI of 1, and 24 h post-infection the cells were washed twice with cold PBS and lysed in 18 ml of lysis buffer (20 mM MOPS-KOH pH 7.4, 120 mM KCl, 0.5% Igepal, 2 mM β-mercaptoethanol), supplemented with 400 unit/ml RNasin (#N2515, Promega) and Complete Protease Inhibitor Cocktail. Cell lysates were incubated on ice for 20 min with gentle mixing every 5 min, and then clarified by centrifugation at 16,000 g for 15 min at 4°C. A 600 μl aliquot of each cell lysate was used to perform total RNA purification using the TRI Reagent LS (#T3934, Sigma). The rest of the cell lysate was incubated for 2 h on a spinning wheel at 4°C with 1.350 ml of StrepTactin High Performance Sepharose beads (#28-9355-99, GE Healthcare). The beads were collected by centrifugation (1,500 g for 5 min at 4°C) and washed twice for 5 min on a spinning wheel with 20 ml of washing buffer (20 mM MOPS-KOH pH 7.4, 120 mM KCl, 2 mM β-mercaptoethanol) supplemented with 400 unit/ml RNasin and Complete Protease Inhibitor Cocktail. A summary of the protocol used to purify MV-N and associated RNA partners from infected cell lysates is depicted in . For RNase protection assay, RNase A/T1 mix reagent (#EN0551, Thermoscientific) was used at different concentrations in the washing buffer: 10 μg/ml of RNaseA with 25 u/ml of RNaseT1; 1 μg/ml of RNaseA with 2.5 u/ml of RNaseT1; 0.1 μg/ml of RNaseA with 0.25 u/ml of RNaseT1. Washed beads were incubated in these RNase A/T1 solutions for 15 min at 37°C and RNase digestion was stopped by addition of TRI Reagent LS.
RNA purification was performed directly from RNA-protein complexes immobilized on StrepTactin Sepharose using TRI Reagent LS. RNA was dissolved in 0.1 ml of DNase-free and RNase-free ultrapure water. The RNAs extracted were analyzed using the nano-drop and Bioanalyser RNA nano or pico kits (#5067-1511 and #5067-1513, Agilent).
Deep Sequencing of RNA
RNA molecules isolated from MV-N/RNA and CH/RNA complexes were treated for library preparation using the mRNA-Seq sample preparation kit (Illumina) according to manufacturer’s instruction. To analyze all RNA species present, the initial poly(A) RNA isolation step was omitted. Briefly, the fragmented total RNA was randomly primed for reverse transcription followed by second-strand synthesis to create double-stranded cDNA fragments. The ends were repaired, an adenine was added to the 3′ end and specific Illumina adapters were ligated. A gel-based size selection at ~200 nucleotides was performed, followed by PCR amplification. Sequencing was performed on the Illumina Genome Analyzer II platform to generate single-end 36 bp reads lacking strand specificity.
Read mapping, expression quantification and statistical analysis
Sequenced reads were cleaned from adaptor sequences and low complexity sequences as described.Citation34 Reads were aligned to the human (hg19, UCSC) and MV Schwarz strain reference genomes (www.ncbi.nlm.nih.gov/nuccore/FJ211590.1) using bowtie (version:0.12.7, options: -a-best -q -m50 -e50 -solexa1.3-qualsCitation35). The first position of each read (taking into account the strandness) was used for statistical analysis using the KNIME software.Citation34 Counts per position where calculated using pileup from samtools.Citation36
Statistical analyses were performed using R version 2.12.1Citation37 and BioConductor (www.bioconductor.org/) packages. Library sizes (total read counts per lane) were normalized according to the TMM scaling factor proposed by Robinson and OshlackCitation13 and implemented in the edgeR package (version 1.8.2).Citation38 They were used to perform a Fisher exact test [as implemented in the sage.test function of the statmod package (version 1.4.8)] to detect positions having significantly different numbers of reads between pairs of conditions. P values obtained from the statistical test were then adjusted for multiple comparisons according to the Benjamini and Hochberg methodCitation39 implemented in the p.adjust function. The normalized library sizes were used to compute the normalized read counts.
RT-PCR detection and cDNA cloning of 5′ copy-back DI-RNAs
The copy-back DI-RNAs were amplified from RNA extracted from MV-N/RNA or CH/RNA samples using two sets of MV primersCitation9: for DI-RNAs, 396 (A: 5′-TATAAGCTTACCAGACAAAGCTGGGAATAGAAACTTCG) and 403 (C: 5′-CGAAGATATTCTGGTGTAAGTCTAGTA) and for the standard genome, 396 (A) and 402 (B: 5′-TTTATCCAGAATCTCAAGTCCGG) ( and ref. Citation9). RT-PCR was performed with SuperScript One-Step RT-PCR with the Platinum® Taq kit (Invitrogen). The PCR-amplified product A-C was cloned into the pTOPO vector (Invitrogen) and sequenced. This plasmid was designated as pTOPO403-396 and possessed the 3′ end of the DI-RNA sequence. The 5′ end of the DI-RNA sequence was PCR-amplified using the MV15875 (5′-ACCAGACAAAGCTGGGAATA) and MV15155 (5′-GCAGCAGATAATTGAATCATCTGTGAGGACTTCAC) primers from pTM2-MVSchw and cloned into the pTOPO vector. This plasmid is designated pTOPO15875-15155. Finally, the 5′ and 3′ ends of the DI-RNA were fused to obtain the full-length MV DI-RNA by BsmBI/BamHI digestion and the final plasmid is designated pTOPO-DIRNA. An additional cloning step was performed to bring the DI-RNA sequence closer to the T7 promoter; the resulting plasmid is called pTOPO-T7DIRNA.
TaqMan RT-qPCR analysis of the 5′ copy-back DI genome and the full-length MV genome
MV DI-RNA RT-qPCR analysis was performed using Applied Biosystem StepOnePlusTMtechnology. RNA was extracted with the TRI Reagent LS before or after affinity chromatography purification on StrepTactin Sepharose. MV DI genome and the full-length genome probes and primers were designed using Primer Express Software (Applied Biosystems). Reactions were performed on 10–1,000 ng RNA using TaqMan RNA-to-Ct 1-Step Kit (#4392938, Applied Biosystems) for one-step RT-qPCR analyses. Reactions were performed in a final volume of 20 μl in the presence of 100 nM TaqMan MV DI-RNA probe (5′-CCCCCGGAACCCTAATCCTGCC), and 250 nM DI-RNA forward (5′-CACTGCCTACCCACGTGACTT) and reverse (5′-TTGCAAATAATGCCTAACCACCTA) primers. The full-length MV genome was amplified with forward (5-TCAGGCATACCCACTAGTGTGAA) and reverse (5′-TGACAGATAGCGAGTCCATAACG) primers in the presence of a specific TaqMan probe (5′-CATCAGAATTAAGAAAAACGTAG) chosen within the H-L intergenic region of the MV genome. β-actin mRNA was amplified as negative control with forward (5-ACCGAGCGCGGCTACAG) and reverse (5′-CTTAATGTCACGCACGATTTCC) primers in the presence of the specific TaqMan probe (5′-CACCACCACGGCCGA). For absolute quantification of MV DI-RNA, MV full-length genome or β-actin, we established standard curves using serially diluted RNAs obtained by in vitro transcription of plasmids encompassing MV DI-RNA, an MV genome fragment or a fragment of β-actin (pTOPO-T7DIRNA, pTOPO-T7MV9119-20as and pTOPO-actin, respectively). The DI-RNA, the full-length genome and the β-actin standard curves were generated by the StepOnePlusTM software system by plotting the Cts against the logarithm of the calculated initial copy numbers. The unknown initial sample copy numbers were then automatically calculated from their Cts, as compared with the RNA standard curves.
Analysis of MV-N/RNA and MV DI-RNA immunostimulatory activity
pTOPO-T7DIRNA was linearized with BamHI before transcription. Short 5′-PPP-bearing RNA molecules 30 nucleotides long were obtained from pCIneo linearized with XbaI before transcription. T7 transcription reactions were performed with a T7 RiboMAXTM Express Large Scale RNA Production System (#P1320, Promega). RNA was purified with RNeasy columns (#74104, Qiagen) and analyzed with a Bioanalyser RNA nano kit (Agilent). Poly(IC) was from Amersham Biosciences (#27-4729-01).
To determine the immunostimulatory activity of the MV-N protein RNA partners, STING-37 cells were plated 1 d before transfection in 24-well (5 × 105 cells per well) plates. Seven ng of RNA isolated on StrepTactin Sepharose or total RNA samples (before One-STrEP-tag purification) were transfected using jetPRIME (#114-15, Polyplus). The cells were lysed 24 h post-transfection and the Firefly luciferase activity was measured using the Bright-Glo Luciferase Assay System (#E2650, Promega) following the manufacturer's recommendation.
Synthetic double-stranded siRNAs were purchased from Applied Biosystems (catalog numbers: s34499 and s34498—targeting MDA5, s223614 and s24143—targeting RIG-I, s33178 and s33180—targeting MAVS and s14354-targeting TPR). A standard protocol of forward siRNA transfection was used. One day before transfection, STING-37 cells were plated in 96-well white microplates (for subsequent luciferase assay), and 5 × 104 cells per well in 0.08 ml DMEM medium with 10% fetal bovine serum, but without antibiotics were seeded. Transfections with siRNAs were performed the following day using Lipofectamine RNAi-MAX (#13778-150, Invitrogen) according to the manufacturer's protocol. For each well, 2 pmol siRNA and 0.2 μl RNAi-MAX transfection reagent combined with 20 μl Opti-MEM serum-free medium (Invitrogen) were added. The cells were transfected 48 h post-siRNA transfection with 6 ng/well of either 1,212-nucleotide-long MV DI-RNA, 5′-PPP-RNA, poly(IC) or mock transfected using jetPRIME. They were lysed 24 h post-transfection and the Firefly luciferase activity in the lysate was measured using the Bright-Glo Luciferase Assay System. Fold induction of Luciferase activity compared with the control (untransfected with stimulatory RNA wells) was established ().
TaqMan RT-qPCR of RIG-I, MDA-5 and MAVS gene expression
Gene expression qPCR analysis was performed using the Applied Biosystem StepOnePlusTM technology. STING-37 cells were transfected with siRNAs targeting MDA5, RIG-I or MAVS and total RNA was extracted using the RNeasy Mini Kit (Qiagen). Expression levels of MDA5, RIG-I and MAVS mRNAs were quantified by one-step real-time PCR using GAPDH mRNA expression as internal control (). Fifty ng of total RNA was amplified with 1 μl custom TaqMan Gene Expression Assays (Hs00920075_m1, for MAVS; Hs01070332_m1, for MDA5; Hs00204833_m1, for RIG-I; Hs99999905_m1 for GAPDH) using TaqMan RNA-to-Ct 1-Strep Kit in accordance with the manufacturers’ instructions. All the measurements were performed in triplicate and analyzed as generated by StepOnePlusTM software system.
| Abbreviations: | ||
| ATU | = | additional transcription unit |
| CH | = | cherry protein |
| ChIP-seq | = | chromatin immunoprecipitation followed by high-throughput sequencing |
| DI genome | = | defective interfering genome |
| IFN | = | interferon |
| HITS-CLIP | = | crosslinking and immunoprecipitation followed by high-throughput sequencing |
| Le | = | genome leader sequence |
| MAVS | = | mitochondrial antiviral-signaling protein |
| MDA5 | = | melanoma differentiation-associated gene-5 |
| MOI | = | multiplicity of infection |
| MV | = | measles virus |
| MV-N | = | measles virus nucleoprotein |
| PAGE | = | polyacrylamide gel electrophoresis |
| RIG-I | = | retinoic acid inducible gene I |
| RNP | = | ribonucleoprotein |
| TMM | = | trimmed mean of M values |
| Tr | = | genome trailer sequence |
Additional material
Download Zip (3 MB)Acknowledgments
We thank Dr Kaoru Takeuchi and Dr Chantal Raboudin-Combe for anti-V antibodies and anti-N antibody, respectively. The authors thank Dr A.-L. Haenni for critical reading of the manuscript, Dr Nicolas Escriou and all the members of the Viral Genomics and Vaccination research Unit for their support and useful discussions.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Financial Disclosure Statement
This work was supported by the Institut Pasteur and the CNRS. A.V.K was supported by a “Bourse Roux” (Institut Pasteur) and by the Agence Nationale de Recherches sur le SIDA et les Hépatites virales (ANRS).
References
- Panganiban AT, Mir MA. Bunyavirus N: eIF4F surrogate and cap-guardian. Cell Cycle 2009; 8:1332 - 7; http://dx.doi.org/10.4161/cc.8.9.8315; PMID: 19342890
- Robb NC, Fodor E. The accumulation of influenza A virus segment 7 spliced mRNAs is regulated by the NS1 protein. J Gen Virol 2012; 93:113 - 8; http://dx.doi.org/10.1099/vir.0.035485-0; PMID: 21918006
- Tanaka T, Kamitani W, Dediego ML, Enjuanes L, Matsuura Y. SARS coronavirus nsp1 facilitates an efficient propagation in cells through a specific translational shutoff of host mRNA. J Virol 2012; 86:11128 - 37; http://dx.doi.org/10.1128/JVI.01700-12; PMID: 22855488
- Vende P, Piron M, Castagné N, Poncet D. Efficient translation of rotavirus mRNA requires simultaneous interaction of NSP3 with the eukaryotic translation initiation factor eIF4G and the mRNA 3′ end. J Virol 2000; 74:7064 - 71; http://dx.doi.org/10.1128/JVI.74.15.7064-7071.2000; PMID: 10888646
- Hawkins RD, Hon GC, Ren B. Next-generation genomics: an integrative approach. Nat Rev Genet 2010; 11:476 - 86; PMID: 20531367
- Baum A, Sachidanandam R, García-Sastre A. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc Natl Acad Sci USA 2010; 107:16303 - 8; http://dx.doi.org/10.1073/pnas.1005077107; PMID: 20805493
- Longhi S. Nucleocapsid structure and function. Curr Top Microbiol Immunol 2009; 329:103 - 28; http://dx.doi.org/10.1007/978-3-540-70523-9_6; PMID: 19198564
- Gubbay O, Curran J, Kolakofsky D. Sendai virus genome synthesis and assembly are coupled: a possible mechanism to promote viral RNA polymerase processivity. J Gen Virol 2001; 82:2895 - 903; PMID: 11714964
- Shingai M, Ebihara T, Begum NA, Kato A, Honma T, Matsumoto K, et al. Differential type I IFN-inducing abilities of wild-type versus vaccine strains of measles virus. J Immunol 2007; 179:6123 - 33; PMID: 17947687
- Whistler T, Bellini WJ, Rota PA. Generation of defective interfering particles by two vaccine strains of measles virus. Virology 1996; 220:480 - 4; http://dx.doi.org/10.1006/viro.1996.0335; PMID: 8661398
- Komarova AV, Combredet C, Meyniel-Schicklin L, Chapelle M, Caignard G, Camadro JM, et al. Proteomic analysis of virus-host interactions in an infectious context using recombinant viruses. Mol Cell Proteomics 2011; 10:M110 - , 007443; http://dx.doi.org/10.1074/mcp.M110.007443; PMID: 21911578
- Devaux P, Cattaneo R. Measles virus phosphoprotein gene products: conformational flexibility of the P/V protein amino-terminal domain and C protein infectivity factor function. J Virol 2004; 78:11632 - 40; http://dx.doi.org/10.1128/JVI.78.21.11632-11640.2004; PMID: 15479804
- Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 2010; 11:R25; http://dx.doi.org/10.1186/gb-2010-11-3-r25; PMID: 20196867
- Lazzarini RA, Keene JD, Schubert M. The origins of defective interfering particles of the negative-strand RNA viruses. Cell 1981; 26:145 - 54; http://dx.doi.org/10.1016/0092-8674(81)90298-1; PMID: 7037195
- Marriott AC, Dimmock NJ. Defective interfering viruses and their potential as antiviral agents. Rev Med Virol 2010; 20:51 - 62; http://dx.doi.org/10.1002/rmv.641; PMID: 20041441
- Strahle L, Garcin D, Kolakofsky D. Sendai virus defective-interfering genomes and the activation of interferon-beta. Virology 2006; 351:101 - 11; http://dx.doi.org/10.1016/j.virol.2006.03.022; PMID: 16631220
- Panda D, Dinh PX, Beura LK, Pattnaik AK. Induction of interferon and interferon signaling pathways by replication of defective interfering particle RNA in cells constitutively expressing vesicular stomatitis virus replication proteins. J Virol 2010; 84:4826 - 31; http://dx.doi.org/10.1128/JVI.02701-09; PMID: 20181705
- Marcus PI, Sekellick MJ. Defective interfering particles with covalently linked [+/-]RNA induce interferon. Nature 1977; 266:815 - 9; http://dx.doi.org/10.1038/266815a0; PMID: 194158
- Yoneyama M, Fujita T. RNA recognition and signal transduction by RIG-I-like receptors. Immunol Rev 2009; 227:54 - 65; http://dx.doi.org/10.1111/j.1600-065X.2008.00727.x; PMID: 19120475
- Vidalain PO, Tangy F. Virus-host protein interactions in RNA viruses. Microbes Infect 2010; 12:1134 - 43; http://dx.doi.org/10.1016/j.micinf.2010.09.001; PMID: 20832499
- Naji S, Ambrus G, Cimermančič P, Reyes JR, Johnson JR, Filbrandt R, et al. Host cell interactome of HIV-1 Rev includes RNA helicases involved in multiple facets of virus production. Mol Cell Proteomics 2012; 11:M111 - , 015313; http://dx.doi.org/10.1074/mcp.M111.015313; PMID: 22174317
- Jäger S, Cimermancic P, Gulbahce N, Johnson JR, McGovern KE, Clarke SC, et al. Global landscape of HIV-human protein complexes. Nature 2012; 481:365 - 70; PMID: 22190034
- Radford AD, Chapman D, Dixon L, Chantrey J, Darby AC, Hall N. Application of next-generation sequencing technologies in virology. J Gen Virol 2012; 93:1853 - 68; http://dx.doi.org/10.1099/vir.0.043182-0; PMID: 22647373
- Routh A, Domitrovic T, Johnson JE. Host RNAs, including transposons, are encapsidated by a eukaryotic single-stranded RNA virus. Proc Natl Acad Sci USA 2012; 109:1907 - 12; http://dx.doi.org/10.1073/pnas.1116168109; PMID: 22308402
- Chi SW, Zang JB, Mele A, Darnell RB. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature 2009; 460:479 - 86; PMID: 19536157
- Rima BK, Duprex WP. The measles virus replication cycle. Curr Top Microbiol Immunol 2009; 329:77 - 102; http://dx.doi.org/10.1007/978-3-540-70523-9_5; PMID: 19198563
- Calain P, Roux L. The rule of six, a basic feature for efficient replication of Sendai virus defective interfering RNA. J Virol 1993; 67:4822 - 30; PMID: 8392616
- Calain P, Curran J, Kolakofsky D, Roux L. Molecular cloning of natural paramyxovirus copy-back defective interfering RNAs and their expression from DNA. Virology 1992; 191:62 - 71; http://dx.doi.org/10.1016/0042-6822(92)90166-M; PMID: 1329337
- Baczko K, Billeter M, ter Meulen V. Purification and molecular weight determination of measles virus genomic RNA. J Gen Virol 1983; 64:1409 - 13; http://dx.doi.org/10.1099/0022-1317-64-6-1409; PMID: 6854275
- Hall WW, Martin SJ. Purification and characterization of measles virus. J Gen Virol 1973; 19:175 - 88; http://dx.doi.org/10.1099/0022-1317-19-2-175; PMID: 4715320
- Combredet C, Labrousse V, Mollet L, Lorin C, Delebecque F, Hurtrel B, et al. A molecularly cloned Schwarz strain of measles virus vaccine induces strong immune responses in macaques and transgenic mice. J Virol 2003; 77:11546 - 54; http://dx.doi.org/10.1128/JVI.77.21.11546-11554.2003; PMID: 14557640
- Giraudon P, Wild TF. Monoclonal antibodies against measles virus. J Gen Virol 1981; 54:325 - 32; http://dx.doi.org/10.1099/0022-1317-54-2-325; PMID: 7288410
- Takeuchi K, Kadota SI, Takeda M, Miyajima N, Nagata K. Measles virus V protein blocks interferon (IFN)-alpha/beta but not IFN-gamma signaling by inhibiting STAT1 and STAT2 phosphorylation. FEBS Lett 2003; 545:177 - 82; http://dx.doi.org/10.1016/S0014-5793(03)00528-3; PMID: 12804771
- Jagla B, Wiswedel B, Coppée J-Y. Extending KNIME for next-generation sequencing data analysis. Bioinformatics 2011; 27:2907 - 9; http://dx.doi.org/10.1093/bioinformatics/btr478; PMID: 21873641
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009; 10:R25; http://dx.doi.org/10.1186/gb-2009-10-3-r25; PMID: 19261174
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al, 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25:2078 - 9; http://dx.doi.org/10.1093/bioinformatics/btp352; PMID: 19505943
- Team RDCR. A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing, 2011.
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010; 26:139 - 40; http://dx.doi.org/10.1093/bioinformatics/btp616; PMID: 19910308
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser A Stat Soc 1995; 57:289 - 300