Abstract
The discovery that Rett syndrome (RTT) is caused by mutation of the methyl-CpG-binding-protein MeCP2 provided a major breakthrough in understanding the neurodevelopmental disorder and accelerated MeCP2 research. However, gene regulation by MeCP2 is complicated. The current consensus for MeCP2 remains as a classical repressor complex, with major emphasis on its role in methylation-dependent binding and repression. However, recent evidence indicates additional regulatory roles, suggesting non-classical mechanisms in gene activation. This has opened the field of MeCP2 research and suggests that the gene targets may not be the usual suspects, that is, dependent only on DNA methylation. Here we examine how chromatin binding and sequence preference may confer MeCP2 functionality, and connect relevant pathways in an active genome. Finding both genomic and proteomic evidence to indicate MeCP2 spliceosome interaction, we consequently discovered broad MeCP2 enrichment of the transcriptome while our focus toward long non-coding RNA (lncRNA) revealed MeCP2 association with RNCR3. Our data may indicate an as-yet-unappreciated role between lncRNA and MeCP2. We hypothesize that ncRNA may mediate chromatin-remodeling events by interacting with MeCP2, thereby conferring changes in gene expression. We consider that these results may suggest new mechanisms of gene regulation conferred by MeCP2 and its interactions upon chromatin structure and gene function.
Introduction
The functional role of MeCP2 has become increasingly complex since its discovery.Citation1,Citation2 The first designation of MeCP2 as a global repressor was initially established through methylated CpG di-nucleotide affinity and the protein’s ability to reduce gene expression, either alone or as part of a complex.Citation3,Citation4 There are at least two categories of methylation-mediated gene suppression. DNA methylation can suppress gene expression itself through directly interfering with the binding of sequence-specific transcription factors.Citation5 Whereas the alternative indirect mechanism involves methyl-CpG-dependent binding proteins, the best characterized of these being MeCP2.Citation3,Citation4 In recent years, there has been a huge increase in the number of characterized repression mechanisms and only a few of them, including MeCP2, can easily be placed into these two categories. Any strict classification of regulation is further complicated by the complexity and diversity of protein–protein interactions involved in the regulation of transcription. Many aspects are likely to contribute to MeCP2 functionality, including A/T nucleotide-enhanced binding,Citation6 high intrinsic disorder,Citation7 and domain interaction, encompassing the recruitment of co-factors.Citation8 Beyond MeCP2’s association with co-repressors, several interactions were thought to be incongruous at first. The association of MeCP2 with the chromatin remodeling complex factors BRM and ATRX,Citation9,Citation10 CREB1,Citation11 RNA, and splicing factorsCitation12-Citation14 are not exclusively repressive and therefore more indicative of a dynamic and multifunctional methyl-CpG-binding protein.
Mutation of MeCP2 in humans is most notably linked to RTT, a severe autism spectrum disorder.Citation15 The predominant neurological phenotype is consistent with high expression levels of MeCP2 in neurons and its effect on nucleosome assembly.Citation16,Citation17 More specifically, reduced histone H1 expression in neurons has been attributed to MeCP2 competition for its nucleosome site.Citation17,Citation18 However, the contribution and targeting of differential gene regulation by MeCP2 still remains poorly characterized. Genome-wide analyses of MeCP2 have revealed a broad binding distribution following patterns of CpG methylation.Citation17,Citation19 Interestingly, the specific binding preference for methylated CpG is only 2–3-fold greater than its unmethylated variant in vitro.Citation20 A review of gene expression analysis in autism spectrum disorders and Rett Syndrome studies suggest the involvement of an abnormal immune response, oxidative stress, altered mitochondrial function, and deregulated protein synthesis, but again provide limited explanation as to why MeCP2 may target these pathways.Citation21-Citation25
Non-coding RNAs (ncRNAs) categorized as either short or long classes are also implicated in transcriptional control.Citation26,Citation27 For example, small interfering RNAs (siRNAs) are thought to participate in sequence-specific chromatin formation guiding the activity of conserved RNAi machinery components.Citation28 This example, and other mechanisms of ncRNA-mediated chromatin assembly are able to regulate gene transcriptionCitation29,Citation30 in the absence of DNA methylation.Citation31 Perhaps the most prominent example of ncRNA regulation in mammals involves the long ncRNA (lncRNA) Xist, which induces silencing of one of the two X chromosomes in females.Citation32 The role of ncRNAs that mediate regulatory control in the brain are yet to be characterized; however, recent discoveries indicate that ncRNAs can mediate regulatory events with specific roles that include the regulation of gene expression,Citation33 as well as chromatin structure and function.Citation31,Citation34-Citation37 Here, our studies extend the understanding of MeCP2 functionality in the brain. Using massively parallel sequencing technologies we examine certain chromatin and sequence attributes, highlight potential targeting associations to relevant pathways, and explore the interaction of MeCP2 with ncRNA. We consider the findings presented here indicate a new role for MeCP2 interacting with ncRNAs to mediate chromatin remodeling changes and gene regulatory events in the brain.
Results
Chromatin fractionation reveals MeCP2 binding in the active genome
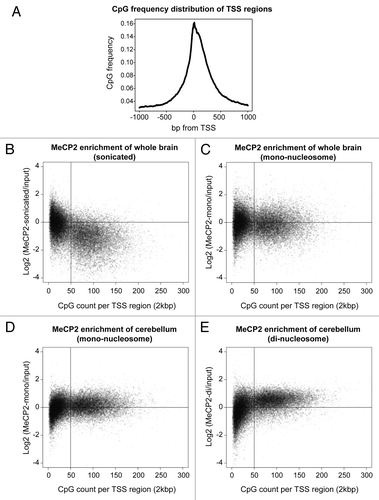
The genome is comprised of a range of varying chromatin structures with diverse functions. In broad terms, chromatin can be characterized either as active with open-euchromatic or inactive with compact-heterochromatic properties, thus enabling fractionation due to their differential resistance to Micrococcal Nuclease (MNase) digestion.Citation38,Citation39 Euchromatin is typically represented in the first fraction, “S1,” as its open structure is readily digestible by MNase. Heterochromatin in contrast has higher resistance to MNase and is represented in a subsequent “S2” fraction.Citation38,Citation39 MeCP2 is largely localized to heterochromatic regions consistent with high levels of methylated DNA and is required for regulating chromatin structure during neuronal maturation.Citation40-Citation42 In order to better understand MeCP2 binding we performed chromatin immunoprecipitation followed by massively parallel sequencing (ChIP-Seq) experiments using sonicated chromatin from whole mouse brain, as well as MNase fractionated S1 euchromatin from both whole mouse brain and the cerebellum alone. In order to account for sonication and digestion biases that may occur due to nucleosome positioning and other factors,Citation43,Citation44 all ChIP-Seq data was analyzed relative to input controls. We designed analysis probes around the transcription start site (TSS) of genes (MM9 assembly) +/− 1000 bp in order to capture peak binding as well as the surrounding distribution of CpG di-nucleotides. The average CpG frequency of these probes rises rapidly when approaching the TSS within +/− 500 bp (). Next, we examined the binding of MeCP2 relative to input material using sonicated chromatin from whole male mouse brain. We show an underrepresentation of TSS regions having more than 50 CpG di-nucleotides, which is likely to reflect genome wide binding of MeCP2 with low CpG density (). In contrast, we observed increased MeCP2 binding at TSS regions with higher CpG di-nucleotide content in the MNase fractionated mono-nucleome selected S1 euchromatin from whole brain (). Indeed, this enrichment was similarly evident in the cerebellum using fractionated mono-nucleosome S1 euchromatin (). MeCP2 enrichment at higher CpG content TSS region probes was further emphasized in S1 di-nucleosome-derived fractions from cerebellum (). Interestingly, there appears to be a critical content threshold of 50 CpG di-nucleotides per 2 kbp TSS region. When exceeding this level, MeCP2 enrichment occurs to a greater extent in S1 fractions and when below, shows enrichment in sonicated whole chromatin. These results highlight relative differences in MeCP2 enrichment between the selective (S1) and non-selective (sonication) techniques, revealing greater association with higher CpG content regions in the active genome as compared with viewing the bulk genome overall.
Figure 1. MeCP2 enrichment of TSS regions in chromatin fractions. Frequency profile plot. (A) Plot shows CpG di-nucleotide frequency over all gene TSS regions +/− 1000 bp. Scatter plots illustrate MeCP2 enrichment of genomic regions designed to the TSS +/− 1000 bp of all genes (MM9 assembly). Individual scatter plots illustrate MeCP2 enrichment relative to input material using sonicated chromatin from whole male mouse brain (B), Mono-nucleosome selected material from S1 fractionated whole male mouse brain (C), and either Mono (D) or Di-nucleosome selected material from S1 fractionated male mouse cerebellum (E). CpG di-nucleotides and ChIP-Seq read base counts occurring in each genomic region were totaled and the enrichment value calculated as Log2 (MeCP2 base count/Input base count).
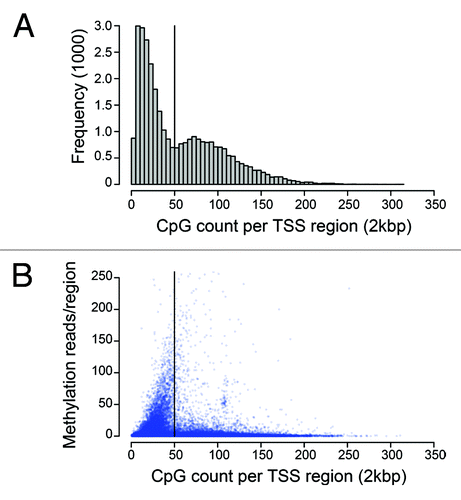
There is a close correspondence of CpG methylation correlated with transcriptional suppression in the existing literature.Citation45 MeCP2 binds strongly to methylated DNA and the predominant model has been a classical co-repressor complex serving to recruit regulatory determinants to suppress gene expression. However, recent experimental evidence has questioned whether the classical view of MeCP2 is an accurate reflection of its role in mediating changes in chromatin structure and transcriptional activity.Citation46 A histogram analysis of CpG content at TSS regions highlights a distribution bias against content approaching approximately 50 CpGs per region, which corresponds with greatest methylation capture (). In addition, the lower methylation levels observed above approximately 50 CpGs, indicates that this threshold content is likely to represent a switch to CpG island status (). Coincidently, this bias point is also the same threshold level that separates MeCP2 enrichment (). As the presence of an un-methylated CpG island near a TSS is commonly associated with actively expressed genes, the MeCP2 enrichment of S1 di-nucleosome material with higher CpG content may indicate chromatin binding that could be associated with gene expression.
Figure 2. TSS region CpG content and methylation distribution. (A) Histogram of TSS (+/− 1000 bp) region CpG content frequency for all genes. (B) Scatter plot of relative methylation level in male mouse cerebellum by TSS region CpG content according to methyl-miner enrichment.
MeCP2 CG-AT4 interaction is enhanced in active chromatin fractions
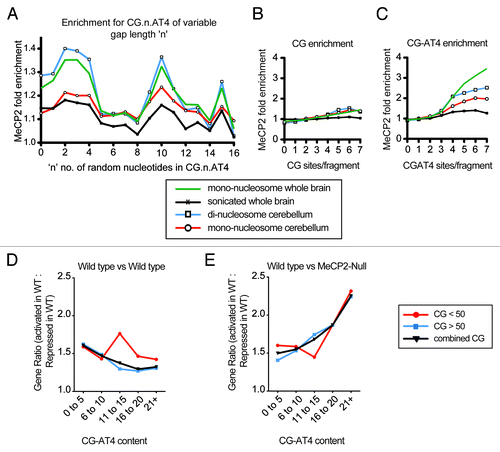
MeCP2 DNA binding is enhanced by the presence of at least four consecutive A or T nucleotides in proximity to the CpG di-nucleotide and represented by the motif CG-AT4.Citation6 To investigate the effect of CpG/AT proximity, we extrapolated 200 bp of sequence for 100 000 ChIP-Seq reads for both input and MeCP2 bound samples and quantified CG-n-AT4 enrichment for “n” representing up to 16 random nucleotides. MeCP2 displayed almost identical enrichment patterns across the sonicated, mono-, and di-nucleosome data sets, with peaks between 0–4, 9–11, and 15 random nucleotides (). The strongest trend was identified in the di-nucleosome data of cerebellum, while data from whole brain sonicated material was the weakest (). The weaker trend in the sonicated as compared to mono-nucleosome whole brain data indicates that CG-AT4 interaction is more frequent in the active genome, while the greater trend in “di” compared to mono-nucleosome data suggests CG-AT4 MeCP2 binding occurs to greater extent within linker DNA (). We also quantified enrichment according to CpG di-nucleotide or CG-n-AT4 motif frequency, with “n” of “0–15.” Greater MeCP2 enrichment was found with increasing CG-AT4 frequency compared with the same number of CpG di-nucleotides (). The greater CG-AT4 enrichment observed in the fractionated data once again supports a role in the active genome, while higher “di” compared to mono-nucleosome cerebellum enrichment again further indicates linker DNA involvement (Fig. 3C). Interestingly, the similar CpG enrichment observed between data sets in Figure 3B are in contrast with CG-AT4 enrichment in Figure 3C, suggesting the positioning of each motif within linker DNA is less critical for CpG as compared to CG-AT4 MeCP2 binding. Hence, the AT repeat should occur on the exposed linker DNA side of the CpG and adjacent nucleosome. These results suggest a preference for CG-AT4 sequences and highlight differential overall contribution to MeCP2 binding between chromatin environments. To examine the relationship between CG-AT4 content and gene expression, we performed comparative RNA-Seq expression analysis of male wild-type and MeCP2-null mouse cerebellum and examined expression by CG-AT4 content within TSS regions. We then considered the ratio of gene expression differences as potentially activated vs. potentially repressed between samples. Grouping genes according to CG-AT4 content revealed an overall reduction of the expression ratio between wild-type samples as CG-AT4 content increased (). This suggests that gene expression is more consistent between samples as CG-AT4 content increases. In contrast, the comparison between wild-type and MeCP2-null cerebellum samples showed a greater proportion of genes that were potentially activated in wild-type with increasing CG-AT4 content (). These trends were also seen if genes were separated into groups with TSS CpG content above or below the 50 CpGs threshold (). Hence, we can infer from these data that the binding of MeCP2 and interaction with CG-AT4 over these specific regions may explain, in part, the differences observed with gene expression.
Figure 3. MeCP2 motif enrichment in genome fractions. Plot s (A–C) indicate MeCP2 fold enrichment of ChIP-Sequencing fragments compared with input for CG.n.AT4 content where there are ‘n’ number of random nucleotides (A), CpG content (B), and CG-AT4 content (C). Reads (100 000) were randomly sampled from sonicated and mono-nucleosome whole brain, mono, and di-nucleosome input and MeCP2 enriched ChIP-Seq libraries. The sampled read sequences were extended to 200bp and quantified for motif feature content. MeCP2 fold enrichment was determined by the ‘MeCP2 enriched reads count’ divided by ‘input reads count’ for sequence reads grouped by feature motif content. Plots (D and E) illustrate the ratio of genes (Activated in wild type: Repressed in wild-type) variation between wild-type samples represented in absolute value (D), and between wild-type and MeCP2-null samples (E). The gene ratio was determined by RNA-Seq analysis of male mouse cerebellum, with grouping according to the CG-AT4 content of regions designed to the TSS +/− 1000 bp of all genes. Plots represent genes with TSS region CG content of below 50 occurences (red), above 50 occurences (blue), or combined CG content genes (black).
Methylation, CG-AT4 TSS content, and MeCP2 differential regulation
The results above indicate the interaction of MeCP2 with active chromatin fractions, which is consistent with prior microarray analysis showing that functional MeCP2 associations are associated with active gene expression.Citation46 To further explore this idea, we employed chromatin immunoprecipitation and methylation sequencing (Methyl-Seq) to compare methyl-CpG profiles and MeCP2 binding across TSS regions. Methylated DNA enrichments were prepared using a Methylminer kit (Invitrogen) with stepwise increase in salt gradients of 350 mM (lower methylation density) and 2000 mM (higher methylation density) to investigate differences associated with methyl-CpG content. In the high salt extractions, only the high CG-AT4 content profile was consistent with methylation-dependent repression (). The profile of high CG-AT4 content with MeCP2-repressed genes indicates stronger methylation levels in comparison to activated genes. This was in contrast to the low CG-AT4 content group in which methylation of MeCP2 repressed genes was lower than the activated gene profile (). This indicates that an increasing number of methylated CG-AT4s in the TSS region resemble the “classical” repressive behavior of MeCP2. However, when we analyzed the density ratio of high- verses low-salt fractions for CpG methylation, a pattern of comparatively higher methylation densities that are up- and downstream of the transcription start sites of repressed genes is revealed, while the activated profiles indicate peak methylation density downstream of the TSS (). Although a similar pattern may be observed between the high and low CG-AT4 content groups, with respect to relative density contributions (high / low methylation signal), a prominent difference remains in the high CG-AT4 group ().
Figure 4. Methylation levels of TSS regions with high CG-AT4 content link MeCP2 differential expression. Traces illustrate Methyl-Miner and MeCP2 ChIP data from cerebellum, over genomic regions aligned to gene TSS +/− 2000 bp. Individual regions were filtered to have at least 50 CpG di-nucleotides and divided into high (> 18 instances) and low (< 7 instances) CG-AT4 content. These regions were further subdivided into groups that were either activated or repressed in wild-type male mouse cerebellum compared with MeCP2-Null according to mRNA-Seq expression analysis. (AandB) display the averaged methylation profiles of TSS region subgroups of high salt (2000 mM) methylminer elution data, while (CandD) shows methylation profiles of high-salt relative to low-salt elution (number of 2000 mM reads divided by 350 mM reads). (E andF) display MeCP2 fold enrichment of di-nucleosome material over input over TSS region subgroups. Profiles of genes activated in wild-type are shown in “green,” and repressed in wild-type as “red.”
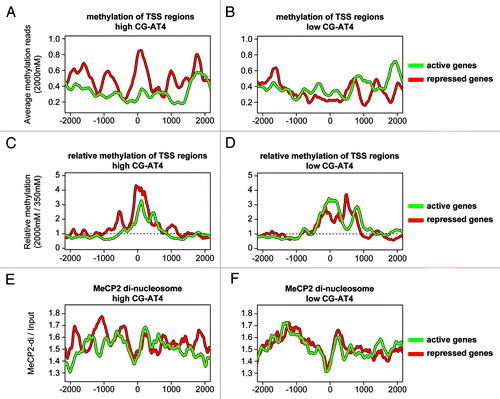
While a clear relationship between differential gene expression, methylation and MeCP2 binding is evident at the TSS, the relationship between high CG-AT4 MeCP2 di-nucleosome enrichment at the TSS is less clear. However, the repressed di-nucleosome binding profile does display differential peaks occurring every 400 to 600 bp upstream of the TSS, which may indicate MeCP2 involvement in repressive positioning of nucleosomes (). The relationship of di-nucleosome binding between differentially regulated genes are less readily apparent in low CG-AT4 profiles. (). While MeCP2 binding profiles between differentially regulated genes show subtle variations, what is readily apparent upstream of the TSS is the lack of MeCP2 di-nucleosome enrichment, which is consistent with nucleosome-free positioning. These data illustrate a relationship between CG-AT4 content and MeCP2 binding status towards specific regulation of gene expression.
CG-AT4 frequency and MeCP2 gene targets
The enrichment of MeCP2 at active sites of chromatin and the relationship with high CG-AT4 content suggests that gene targets in the brain share these genomic features that regulate MeCP2-mediated gene expression. To investigate this, we used a bioinformatics approach and screened CG-AT4 content in both mouse and human genomes as a potential marker for MeCP2 interaction. Sequences were extracted for regions 1 Kbp +/− of the TSS, while counting CpG and CG-AT4 motif content. Approximately 3000 of the highest or lowest genes according to TSS motif content were analyzed for gene pathway association (). KEGG pathways analysis of high CpG content genes associated largely with cancer, while both low CpG and low CG-AT4 content genes link to olfactory and compound metabolic pathways. However, while there were unique relationships with high CpG content, there were strong relationships readily apparent when quantified for the high occurrence of CG-AT4 motifs. For example, inspection of our large data sets demonstrates that for high CG-AT4 content genes, there are significant relationships of pathway analysis demonstrating links to neurodegenerative disease and pathways associated with MeCP2 (). These included oxidative phosphorylation, Huntington’s, Alzheimer, and Parkinson disease, which share several genes relating to mitochondrial function and encode respiratory complex subunits. Previously, mitochondrial abnormalities relating to loss of MeCP2 function were linked to respiratory complex III by the UQCRC1 subunit.Citation25 Significant relationships of this type were evident and while a clear association between CG-AT4 content and gene pathways implicated in neurodegenerative pathways is prominent, the potential interaction of MeCP2 with spliceosome components at the gene level was intriguing. This is because it expands upon recent experimental results by Young et al. that highlight MeCP2 involvement with RNA, its splicing and interaction with RNA proteins.Citation14 The ribosomal pathway may also be linked to RTT and neurodegenerative disease through aberrant protein synthesis associated with these conditions.Citation24,Citation47 A comparison of high CG-AT4 motif content with high MeCP2 di-nucleosome binding showed that a number of pathways are shared between the differentially selected genes. Once again, ribosome, spliceosome, and neurodegenerative pathways, including Huntington’s disease, were identified (). Additionally, a comparison of high CG-AT4 content genes from either high or low MeCP2 binding groups revealed that the previously observed pathways are associated with high MeCP2 enrichment (). These results illustrate that CG-AT4 sequences in context to chromatin have the potential to regulate gene expression by targeting MeCP2 to the genome.
Table 1. Gene pathway analysis of motif content in TSS regions
Table 2. Gene pathway analysis: high CG-AT4 content vs high MeCP2 Di-nucleosome binding
Table 3. Gene pathway analysis: low vs high MeCP2 Di-nucleosome binding
MeCP2 co-factor interaction highlights the spliceosome
Our experiments have examined MeCP2 functionality in terms of genome-wide and sequence-specific binding. The enrichment of MeCP2 at CG-AT4 sequences and gene pathways associated with ribosome and spliceosome as well as predominant binding of MeCP2 with di-nucleosomes suggest interactions with RNA networks. To examine further the relationship between MeCP2 and its involvement with RNA, soluble nuclear extracts from whole mouse brains were prepared, followed by endogenous MeCP2 co-immunoprecipitation (Co-IP) with and without RNase treatment. The enriched protein samples were then processed for protein identification by mass spectrometry (MS). Fewer proteins were detected within the RNase-treated sample () in agreement with the RNA dependency of large MeCP2 complexes.Citation14 Analysis of both the RNase-treated and -untreated samples identified proteins having DNA- and RNA-dependent functions. Consistent with the above results, MS protein fingerprinting also showed a predominant association of spliceosomal components, reiterating the association of MeCP2 with RNA (). Western analysis of MeCP2 Co-IP material confirmed the interaction with the spliceosome component PRP8, the RNA-dependent interactions of TOP2b and DHX9, as indicated by MS, as well as interaction with BRM and BRG1 (). These results indicate a RNA dependent class of MeCP2 interactions.
Figure 5. MeCP2 co-immunoprecipitation confirms M/S identified proteins and shows RNA dependance. Soluable mouse brain nuclear extract was pre-treated with either 0, 10, 30, or 100 µg/mL RNase A before Co-immunoprecipitation using endogenous MeCP2. The precipitated proteins were resolved by SDS-PAGE for mass spectrometry analysis and western blot confirmation. Proteins listed in (A) were identifiable through mass spectrometry analysis of MeCP2 Co-IP material with or without RNase treatment as indicated. Western blot confirmations (B) were performed for mass spectrometry identified proteins where effective antibodies were available. These proteins included TOP2b-DNA topoisomerase, DHX9 RNA helicase, and PRP8 RNA splicing factor. Western blots were also examined for the SWI/SNF components BRM and BRG-1, previously shown to interact with MeCP2.

Functional correlation suggests greater roles between MeCP2 and RNA
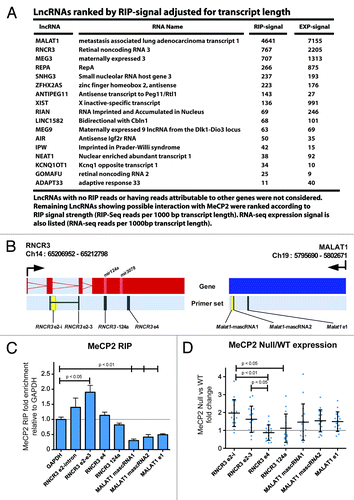
The identification of MeCP2 protein interacting with RNA-based proteins was surprising given that previous studies focused on regulatory complexes associated with gene transcription.Citation48 Since MeCP2 interacts with RNA processing proteins, we screened for MeCP2-associated RNA using a modified RNA-ChIP-Seq protocol (RIP-Seq). MeCP2-immunopurified complexes from fixed, sonicated wild-type mouse cerebellum were DNase treated, followed by massively parallel sequencing of enriched RNA transcripts. RIP experiments were also performed using MeCP2-null cerebellum but did not yield detectable levels of enriched RNA for use in sequencing library generation. The sequencing data indicates that MeCP2 associates with many RNAs, with the strongest signals originating from those genes with greatest expression. Similar functional groups were found in pathway analysis of high RIP-Seq signal transcripts as observed in analyses of both high di-nucleosome ChIP binding and high CG-AT4 TSS content (). However, pathway similarity was lost if the analysis of RIP-Seq was performed relative to expression level (). Since RNA binding domains exist outside of the methyl-CpG binding domainCitation12 and given high MeCP2 expression in the brain, binding might be proportional to mRNA levels. While specific interactions with RNA have not been associated with MeCP2, preference for double stranded RNA has been reported.Citation12 We therefore directed our focus toward long non-coding RNAs (lncRNAs) as they frequently form secondary structures and have been implicated with chromatin structure formation.Citation49 While a number of lncRNAs are likely to interact with MeCP2, we selected the high RIP-Seq signal transcripts RNCR3 and MALAT1 for validation (). Using qRT-PCR relative to GAPDH and input, we observed higher relative enrichment of RNCR3 compared with MALAT1, with greatest enrichment over RNCR3 exons 2 and 3 (). While there is a clear relationship between MeCP2 associated with the exons of RNCR3, the relationship between MeCP2 and RNCR3-124a, which encodes the mir124a, was less evident indicating that the interaction appears to be predominantly exonic with sequence specificity. In contrast this interaction is significantly reduced for MALAT1 in the cerebellum, which suggests the higher RIP-Seq signal for Malat1 may be attributed to greater transcript copy number. Next, we considered the possibility that lncRNA binding to MeCP2 could differentially regulate RNCR3 and MALAT1 gene expression. Comparative expression between wild-type and MeCP2-null male mouse cerebellum revealed greater expression of RNCR3 exons 2 and 3 compared with exon 4 in null tissue, but unremarkable change in MALAT1 (). The variation in RNCR3 exonic expression in the absence of functional MeCP2, suggests abnormal transcription or splicing events between exons two to three and the forth exon of the RNCR3 transcript. Hence, it is likely that MeCP2 is required for the normal regulation of these processes. These results are consistent with MeCP2-associated lncRNA interactions and further implicate MeCP2 with RNCR3.
Table 4. High and Low RIP signal pathways analysis
Figure 6. MeCP2 RIP sequencing of LncRNAs and assessment of RNCR3 and MALAT1. (A) List of MeCP2 RIP captured LncRNAs ranked by RIP-sequencing reads per 1000 bp of transcript length. LncRNAs having either no RIP reads or reads that were indistinguishable from the enrichment of an overlapping genomic transcript were not considered in this list. (B) Expression and RIP primer sets were designed over RNCR3 and MALAT1, chromosomes 14 and 19 respectively as shown: TSS is indicated by directional arrows, gene structure shows either red or blue exon regions according to direction. Overlapping primer sets occur in yellow. Positioning of micro RNAs are marked in the gene structure. (C) MeCP2 RIP fold enrichment adjusted to input and relative to GAPDH, error bars indicate SEM. (D) Expression data shows distribution, mean, and standard deviation of RT-PCR differential expression analysis between wild-type and MeCP2-null mouse cerebellum. RIP data were analyzed by t-test compared with GAPDH, and expression data analyzed by ANOVA. Significant P values are as indicated.
Discussion
The data presented here identifies a role for MeCP2 in the regulation of gene expression that highlights physiological targets in the brain. The binding of MeCP2 to CG-AT4 is enhanced in active chromatin fractions. We also observe MeCP2 interactions with long non-coding RNAs. The identification of MeCP2 association with the splicesome at multiple levels was also surprising given that previous studies have demonstrated interactions that focus on DNA binding transcription factors and core machinery that serve to alter chromatin structure and regulate gene expression.
The first glimpse into the mechanism of methylation mediated silencing by MeCP2 came in 1998 from the seminal papers from the groups of Adrian Bird and Alan Wolffe, when they reported the three-way connection between DNA methylation and gene silencing was associated by the modification of chromatin.Citation3,Citation4 The discovery that MeCP2 binds a co-repressor complex that includes mSin3A and histone deacetylases paved the way to a molecular mechanism between MeCP2 and chromatin.Citation50,Citation51 A large body of evidence followed these reports exploring the control of gene expression by MeCP2.Citation52,Citation53 Coordinated by DNA methylation and given its preference for the 5-methyl-cytosine moiety, MeCP2 was largely associated with transcriptional repression and chromatin remodeling.Citation9,Citation10 Although specific MeCP2 targets have been sought, recent studiesCitation17,Citation19 and our current data illustrate more widespread genome-wide interactions. While CG-AT4-enhanced binding has been considered for some time,Citation6 CG-AT4 frequency has not been examined for targeting of MeCP2 to the genome. In this regard, we show a link of MeCP2 activity to neurodegenerative disease, energy production, protein synthesis, and RNA splicing pathways, which are molecular processes thought to be involved in RTT and autism pathophysiology.Citation22-Citation25 The specific regulation of genes by MeCP2 requires additional experimental investigation, but we find that CG-AT4 frequency and methylation status can account for some of the variability. Commonality between MeCP2 di-nucleosome enrichment and high CG-AT4 pathways suggest MeCP2 competition with histone H1 for linker DNA in brain. In support of this, histone H1 was shown to be enriched exclusively in di-nucleosome material from Alzheimer patients.Citation54 This study clearly implicated H1 with neurodegenerative disease, while highlighting the importance of chromatin context. The specific affinity of MeCP2 for the chromatin template over a region may define its ability to compete with histone H1, which in turn (de)stabilizes chromatin structure and altering gene transcription. Indeed, the effectiveness of MeCP2 to alter chromatin structure was recently shown in mouse models expressing mutations at amino acids 270 and 273 of the MeCP2 protein.Citation55 This mutation resembles the severe phenotype seen in Rett individuals, the authors of the study identifying a 250 amino acid stretch homologous to HMGA1that encodes three AT-hook-like domains that alter ATRX localization. These results suggest the AT-hook of MeCP2 is important to maintain chromatin structure.
The multifaceted DNA binding properties of MeCP2 were also recently expanded to 5-hydroxymethyl-CpG (5hmC) sites in the brain to further highlight its interaction with euchromatin could regulate genome wide changes in gene expression.Citation56 Although the current consensus for MeCP2 remains as a “classical” repressor complex with the major emphasis of research on its well-characterized role with respect to methylation-dependent binding,Citation57 recent studies now show additional regulatory roles that are more suggestive of a “non-classical mechanism” of regulation mediating gene activation.Citation11,Citation46 An elegant solution to the long standing relationship that MeCP2 recognizes unmethylated DNA and regulates euchromatin was recently shown in the brain.Citation56 The high levels of 5hmC in neuronal genomes, specifically in euchromatin of mature cerebellar cells was associated with MeCP2 binding which was disrupted by the Rett mutation, R133C. Interestingly, 5hmC enrichment at active genes were depleted for 5mC at these regions which were consistent with strong affinity of MeCP2 at these sites. These findings now open the field of MeCP2 research and suggests that the gene targets may not be the usual suspects, i.e., dependent on DNA methylation. Clearly, these recent experimental findings demonstrate how MeCP2 recognizes the genome and is targeted to chromatin is as important as the gene under regulation.Citation58,Citation59 Indeed, the identification of MeCP2 enrichment at 5hmC sites and its relationship with active euchromatin suggests the reevaluation of its binding properties. The binding of MeCP2 on 5mC and 5hmC with similar affinities could explain gene activation as well as transcriptional suppression in animal models that either lack or overexpress MeCP2.Citation11,Citation60
Consistent with recently published data, we observed broad genomic enrichment,Citation17 but also identified unique patterns of MeCP2 binding between different genomic fractions. While a clear relationship exists between differential gene expression and DNA methylation, as well as di-nucleosome binding and high CG-AT4 content,, this relationship becomes less readily apparent when considering low CG-AT4 TSS regions. High CG-AT4 pathways analysis suggests neurodegenerative disease associated with MeCP2 function. We also identified pathways associated with the spliceosome and ribosome with MeCP2. In addition, our mass spectrometry results also implicate the participation of MeCP2 with RNA regulation and the spliceosome. This would suggest a model of regulation mediated by MeCP2 in the regulation of RNA networks and could account for MeCP2 interaction with the RNA binding protein YB-1.Citation14 Indeed, the alternative splicing of RNA transcripts in the brain is critical to regulating gene expression patterns and neuronal plasticity.Citation61 We have identified several proteins by mass spectrometry associated with RNA remodeling and the spliceosome such as DHX-9 and PRP-8, respectively. The ATP-dependent RNA helicase, DHX9 functions in the unwinding of double-stranded RNA and DNA-RNA complexes.Citation62 Its gene, DHX-9, belongs to a family of DEAH-box RNA-helicases characterized by the Asp-Glu-Ala-His motif and is implicated in RNA metabolism and transcriptional activation.Citation63 We found that DHX-9 is an RNA-dependent MeCP2-interacting protein by mass spectrometry and this was validated by protein co-immunoprecipitation in nuclear extracts. Given the abundance of MeCP2 in the brain, its enrichment with heterochromatin and involvement in maintaining chromatin structure, we hypothesize the interaction with DHX-9 could also regulate RNA and DNA-RNA chromatin complexes. Although the mechanism by which MeCP2 cooperates with RNA helicases remains to be determined, this seems probable based on mass spec-protein identification. We also found MeCP2 interacting with members of the DEAD-box family characterized by the conserved motif Asp-Glu-Ala-Asp such as DDX-proteins.Citation64 For example, the ATP-dependent RNA helicase, DDX5 is involved in alternative regulation of mRNA splicing mediated by DNA- and chromatin-binding protein factors.Citation65 A central component of the spliceosome, the PRP-8 protein is a component of U2- and U12-dependent spliceosomes and essential in pre-mRNA splicing events.Citation66 PRP-proteins also interact with SWI/SNF enzymes to coordinate pre-mRNA splicing with chromatin remodeling events involved in transcriptional regulation.Citation67 The relevance of these findings to MeCP2 suggests a possible role in the regulation of chromatin structure through interaction with components of RNA splicing.
Alternative splicing is observed in higher eukaryotes to diversify transcriptomes and the serine/arginine (SR) splicing factors are engaged in regulating neuronal gene expression. Long non-coding RNAs such as MALAT1 interact with pre-mRNA splicing factors to regulate alternative splicing.Citation68,Citation69 In this study, we show interacting RNAs by screening for MeCP2 using a modified RIP-Seq technique. MeCP2-immunopurified complexes identified RNA transcripts enriched for several lncRNAs, including MALAT1 and RNCR3. Although a direct mechanism by which lncRNAs could regulate MeCP2 binding remains to be determined, we speculate that ncRNAs such as RNCR3 guide MeCP2 to chromatin to regulate gene structure and function, and that dsRNAs may also act to buffer MeCP2 chromatin binding through displacement. To our knowledge, this is perhaps the first demonstration of context preference as well as the association of MeCP2 with lncRNA that may mediate targeting of regulatory functions. Recently, an RNCR3−/− mouse model was created to study MIR-124A sourced from the RNCR3 lncRNA.Citation70 Intriguingly, RNCR3−/− mice display hind limb clasping, reduced brain weight, and other characteristics that are reminiscent of those seen in MeCP2-null mice, which model the clinical features of RTT.Citation70 Furthermore, both MeCP2 and RNCR3 are required in normal GABAergic neuron function and development.Citation71,Citation72 Given the high capacity of ncRNAs to form secondary structures and the ability of double stranded RNA to displace MeCP2 from bound methylated DNA,Citation12 this previously unknown connection to RNCR3 offers an alternative mechanism to MeCP2 gene regulation. In addition to its participation in RNA splicing, the interaction of MeCP2 with ncRNAs extends the mechanism of gene regulation by targeting MeCP2 to chromatin, beyond the context of CpG methylation and CG-AT4. The association of ncRNA provides a new entry point in understanding the MeCP2 regulatory mechanisms as well as providing some insight of how the ncRNA assembly may guide MeCP2 to chromatin. The identification of such MeCP2 gene targets could be achieved by examining contextual relationships of chromatin and their role in the function of lncRNAs and other RNA molecules.
Based on the findings presented here, the experimental data allow us to reassess MeCP2-mediated gene regulation. We hypothesize MeCP2-interacting ncRNAs may mediate chromatin remodeling changes and gene regulatory events in the brain. Although we do not fully appreciate the relative importance of the spliceosome proteins and the MeCP2 interacting long non-coding RNAs, the results presented allow reinterpretation of long-standing questions relevant to our understanding of gene regulation as well as providing insight for MeCP2 gene targets. Not only is this of direct therapeutic relevance to Rett syndrome, but will also be important for our understanding of new mechanisms of gene regulation conferred by MeCP2 interacting ncRNAs that influence chromatin structure and function.
Methods and Materials
Animals
Brain tissue for sequencing experiments was obtained from wild-type (mixed 129/C57BL6 background) or symptomatic MeCP2-null (Mecp2tm1Tam) male mice.Citation73 Mass spectrometry analysis was performed using brains collected from swiss-webster mice (Pel Freeze Biologicals).
Antibodies
MeCP2 (9317, Sigma), Prpf8 (ab51366, Abcam), DHX9 (NB110-40579, Novus Bio), Histone H1 (05-457, Millipore), TOP2b (NB100-40842, Novus Bio), BRM (ab15597, Abcam), BRG-1 (Dr Said Sif).
Soluble nuclear protein extraction
All buffers and steps were kept on ice unless otherwise stated. All buffers contained 1x complete protease inhibitor cocktail tablets (Roche Diagnostics). Nuclear extracts were prepared from whole mouse brains (Pel-Freez Biologicals). Five brains were dounce homogenized in PBS and pelleted (2 min, 1500 g). The homogenized tissue was resuspended in 10 mL low-salt buffer (10 mM HEPES, 15 mM KCl, 0.2 mM EDTA, 0.1 mM EGTA, 0.5 mM DDT, 0.5 mM spermidine, 0.15 mM spermine, 0.075% NP40) for 30 min at 4 °C. Nuclei were isolated by mixing one part sucrose buffer (50 mM HEPES, 10 mM KCl, 0.2 mM EDTA, 0.1 mM EGTA, 0.5 mM DDT, 0.5 mM spermidine, 0.15 mM spermine, 75% sucrose) with two parts low-salt homogenate and centrifugation (30 s, 8000 g). The isolated nuclei pellet was resuspended with 2x the pellet volume of high-salt buffer (50 mM HEPES, 450 mM KCl, 0.2 mM EDTA, 2 mM EGTA, 0.5 mM DDT, 1 mM MgCl, 25% gylcerol), and incubated 5 min at 4 °C. Soluble nuclear protein extract (supernatant) was recovered by centrifugation for 15 min at 18 000 g, then snap frozen and stored at -80 °C.
Co-immunoprecipitation
For each immunoprecipitation, RNase treated (100 µg RNase A [Sigma] for 30 min at 4 °C unless otherwise stated) or non-RNase-treated soluble nuclear extract was used. A 150 µL nuclear extract, adjusted to contain 1% BSA, was precleared twice (2 × 45 min), using 20 µL packed volume protein A Sepharose beads (GE Healthcare) each time. Precleared extracts were incubated for 1 h with MeCP2 antibody (Sigma), before adding 20 µl packed volume of protein A Sepharose beads blocked with 1% BSA and a further 2 h incubation at 4 °C. Bead complexes were washed five times with washing solution (1 × PBS, 1% Triton X-100), before boiling in 40 µl elution buffer (0.1 M glycine pH 2.5, 50 mM DTT). Precipitated proteins were size fractionated by SDS PAGE, and alternately processed for mass spectrometry or transferred to PVDF membrane for examination by western blotting using standard techniques.
Mass spectrometry
SDS PAGE gels of MeCP2 co-IP proteins were stained with coomassie blue R-250 (Amresco). Protein bands were excised and initially cleaned by sonication in 10 mM Tris for 5 min. Standard techniques for in-gel protein digestion followed, involving sequential dehydration (50% acetonitrile, 25 mM ammonium bicarbonate) and rehydration (100 mM ammonium bicarbonate) treatments to further clean and remove coomassie staining from gel pieces. Cysteine residues were reduced (10 mM DTT, 100 mM ammonium bicarbonate, 60 °C for 1 h) and carbamidomethylated using iodoacetamide (55 mM, 20 min). Treated gel pieces were further washed (100 mM ammonium bicarbonate) and finally with water before drying by speed vac (Thermo Scientific). Dried gel pieces were hydrated with digestion solution (0.05 µg/mL trypsin (Promega) in 25 mM ammonium bicarbonate) on ice for 30 min, excess digest solution was removed by washing with and final suspension in 10 µL digest buffer (25 mM ammonium bicarbonate). Trypsin digest of proteins continued for 3 h at 37 °C, and left at 4 °C overnight. A 0.6 µL aliquot of MALDI matrix (α-cyano-4-hydroxy cinnamic acid, 3 mg/ml in acetone/ethanol [1:2, v/v]) was spotted to an anchorchip target plate (Bruker Daltonics) with 0.5 µL acidified protein digest (0.1% TFA), and examined by MALDI-TOF using an Autoflex II mass spectrometer, Flex, and biotools analysis software (Bruker Daltonics). The mass range (900–3000 mhz) of the mass spectrometer was internally calibrated using autolytic peaks of trypsin. Peptide mass lists of digests were compared with the protein database SwissProt using the mascot search algorithm (Matrix Science, http://www.matrixscience.com). Typical search parameters were as follows: mass tolerance, 0.25 Da; missed cleavages, 2; enzyme, trypsin; fixed modifications, carbamidomethylation; variable modification, Oxidation (M); taxonomy, Mus musculus. Protein ID matches with probability based Mowse scores of at least 82 were recorded (P < 0.0001).
Chromatin-sonication
The frozen brain of one mouse was homogenized in 5 mL of 1% formaldehyde PBS solution using a 10 mL dounce (Wheaton). Cross-linking of the homogenate solution continued for a total of 10 min at room temperature (RT) with rotation from the start of homogenization (Note: within the last 2 min the homogenate was centrifuged [2 min, 1500 g, RT]). Formaldehyde cross-linking was halted at exactly 10 min by addition of 5 mL 125 mM glycine PBS solution to the pellet and a further 10 min incubation at RT with rotation. The quenched solution (200 µL) was aliquoted into 5 tubes (1.5 mL, Eppendorf), pelleted, and re-suspended in 250 µL lysis buffer (1% SDS, 10 mM EDTA, and 50 mM Tris-HCl pH8.0). Lysed material was sheared to between 100 and 500 bp using a waterbath sonicator (Diagenode). For each aliquot 200 µL of cleared sonicate was collected and added to 1.8 mL ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl pH8.0, and 167 mM NaCl) with 40 µg RNase A (Sigma) and incubated for 30 min at 4 °C with rotation and finally pooled.
Chromatin-MNase fractionation
All buffers and steps were kept on ice unless otherwise stated. The brain or cerebellum of one mouse was isolated and homogenized in 5 mL of 1% formaldehyde PBS solution using a 10 mL dounce (Wheaton). Cross-linking of the homogenate solution continued for a total of 10 min at room temperature (RT) with rotation from the start of homogenization (Note: within the last 2 min the homogenate was centrifuged [2 min, 1500 g, RT]). Formaldehyde cross-linking was halted at exactly 10 min by addition of 5 mL 125 mM glycine PBS solution to the pellet and a further 10 min incubation at RT with rotation. The formaldehyde quenched pellet was isolated (2 min, 1500 g, RT), and 5 mL of nuclear isolation buffer (15 mM Tris-HCL pH 7.5, 60 mM KCl, 0.5 M sucrose, 0.25 mM EDTA, 0.125 mM EGTA, 1 mM DTT, 0.5 mM PMSF, 0.15 mM spermidine, 0.15 mM spermine, 0.2% NP-40, and 1X complete proteinase inhibitor [Roche]) was added and incubated for 5 min at 4 °C with rotation. The solution was equally divided between four 1.5 mL tubes (Eppendorf) and the nuclear pellet isolated by centrifuge (1 min, 5000 g, 4 °C). Each pellet was washed briefly with 200 µL MNase digestion buffer (20 mM Tris-HCl, 70 mM NaCl, 20 mM KCl, 5 mM MgCl, 3 mM CaCl, 0.5 mM PMSF, and 1X complete proteinase inhibitor [Roche]) and centrifuged (1 min, 4000 g, 4 °C). Each washed pellet was re-suspended in 400 µL of MNase digestion buffer and rested on ice, before adding 250 units micrococcal nuclease (Worthington) and allowing digestion for 8 min. Digestion was stopped by addition of 16 µL MNase stop solution (125 mM EDTA, 125 mM EGTA). The digest was centrifuged (5 min, 5000 g, 4 °C) and the fixed s1 chromatin fraction (supernatant) from each tube collected and pooled. RNA was digested from collected S1 samples by RNase treatment at the rate of 100 µg RNase A (Sigma) per mL of sample for 30 min at 4 °C with rotation.
Chromatin immunoprecipitation
ChIP was performed on both sonicated and MNase fractionated chromatin. All buffers and steps are kept on ice unless otherwise stated. A 40 µL aliquot was retained as Input material. For MNase fractionated chromatin 100 µL sample was aliquoted into each of 16 tubes (1.5 mL, Eppendorf), along with 20 µL Protein A Dynabeads (Invitrogen) and 400 µL ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl pH8.0, and 167 mM NaCl). For sonicated material 500 µL diluted sample was added to each of 16 tubes with 20 µL Protein A Dynabeads (Invitrogen). For each experiment, an additional set of 16 tubes were set up in parallel containing 500 µL ChIP dilution buffer and 20 µL Protein A Dynabeads, with 2.5 µg of antibody added to eight tubes only. All tubes were placed at 4 °C with rotation for 1 h allowing pre-clearing of sample and pre-binding of antibody to beads. The pre-cleared chromatin was separated from dynabeads using a magnetic rack (Invitrogen) and transferred to the parallel set of either pre-bound or no-antibody bead containing tubes of which solution was discarded. Antibody and no-antibody control tubes were then left over night to at 4 °C with rotation in order to precipitate chromatin. The following day chromatin bound beads were washed twice consecutively with 0.5 mL each time of the following buffers in order; ChIP dilution buffer, Low Salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH8.0, and 150 mM NaCl), High Salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH8.0, and 500 mM NaCl), LiCl buffer (0.25 M LiCl, 1% Tergitol, 1% deoxycholic acid, 1 mM EDTA, and 10 mM Tris-HCl pH 8.0), and TE buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA). After washes, beads were transferred to a new set of tubes using 0.5 mL of TE buffer with 0.01% SDS and the solution discarded. To each tube of antibody or no-antibody control beads, 100 µL of RT ChIP elution buffer (20 mM Tris-HCl pH 7.5, 5 mM EDTA, 50 mM NaCL, and 1% SDS) and 40 µg protinase K (P4850, Sigma) was added. A360 µL aliquot of ChIP elution buffer and 120 µg protinase K was added to the retained input material of 40 µL. The elutions of ChIP material and Input were incubated at 62 °C in a thermomixer (Eppendorf) at 1400 rpm for 2 h to reverse cross-links. Beads were then removed and ChIP samples pooled into Antibody bound, No-Antibody control, and Input. An equal volume of Phenol: Chloroform: Isoamyl Alcohol was added to each sample, then vortexed, and rotated at RT for 10 min before centrifugation (15 min, 18 000 g, RT). DNA was purified from the aqueous phase using the Nucleospin extract II kit (Macherey-Nagel) as per manufacturer instructions. Purified DNA was used to make genome sequencing libraries or in RT-PCR.
RNA immunoprecipitation
The frozen cerebellum of one mouse was homogenized in 5 mL of 1% formaldehyde PBS solution using a 10 mL dounce (Wheaton). Cross-linking of the homogenate solution continued for a total of 10 min at room temperature (RT) with rotation from the start of homogenization (Note: within the last 2 min the homogenate was centrifuged [2 min, 1500 g, RT]). Formaldehyde cross-linking was halted at exactly 10 min by addition of 5 mL 125 mM glycine PBS solution to the pellet and a further 10 min incubation at RT with rotation. The quenched solution (200 µL) was aliquoted into 5 tubes (1.5 mL, Eppendorf), pelleted, and re-suspended in 300 µL RNA lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl pH8.0, 0.5 mM PMSF, 60 units RNase inhibitor (Applied Biosystems), and 1X complete proteinase inhibitor [Roche]). Lysed material was sheared to between 100 and 500 bp using a waterbath sonicator (Diagenode). For each aliquot 250 µL of cleared sonicate was collected and added to 1.8 mL ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl pH8.0, and 167 mM NaCl), 200 µL was retained as Input material. The diluted material (500 µL) was added to each of 4 tubes with 20 µL Protein A Dynabeads (Invitrogen). For each experiment, an additional set of 4 tubes were set up in parallel containing 500 µL ChIP dilution buffer and 20 µL Protein A Dynabeads, with 2.5 µg of MeCP2 antibody added to two tubes only. All tubes were placed at 4 °C with rotation for 1 h allowing pre-clearing of sample and pre-binding of antibody to beads. The pre-cleared chromatin was separated from dynabeads using a magnetic rack (Invitrogen) and transferred to the parallel set of either pre-bound or no-antibody bead containing tubes of which solution was discarded. Antibody and no-antibody control tubes were then incubated 2 h at 4 °C with rotation to precipitate RNA complexes. RNA complex bound beads were washed twice consecutively with 0.5 mL each time of the following buffers in order; ChIP dilution buffer, Low Salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH8.0, and 150 mM NaCl), High Salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH8.0, and 500 mM NaCl), LiCl buffer (0.25 M LiCl, 1% Tergitol, 1% deoxycholic acid, 1 mM EDTA, and 10 mM Tris-HCl pH 8.0), and TE buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA). After washes, beads were transferred to a new set of tubes using 0.5 mL of TE buffer with 0.01% SDS, and the solution discarded. To each tube of antibody or no-antibody control beads, 100 µL of RIP elution buffer (20 mM Tris-HCl pH 7.5, 5 mM EDTA, 50 mM NaCL, 1% SDS, 20 units RNase inhibitor, and 40 µg protinase K [P4850, Sigma]) was added. Forty units RNase inhibitor and 80 µg protinase K was added to the 200 µL of diluted input material. The elution samples and input were incubated at 62 °C in a thermomixer (Eppendorf) at 1400 rpm for 2 h to reverse cross-links. Beads were then removed and ChIP samples pooled into Antibody bound, No-Antibody control and Input. An equal volume of Chloroform was added to each sample, then vortexed, and rotated at RT for 10 min before centrifugation (15 min, 18 000 g, RT). RNA was purified from the aqueous phase using the RNAeasy Minelute kit with DNase pre-digestion (Qiagen) as per manufacturer instructions. RNA was eluted in 30 µL nuclease free water, and used to make RNA sequencing libraries or in RT-PCR analysis.
Methylated DNA enrichment
Male mouse cerebellum DNA (100 µg) was sonicated to an average length of approximately 300 bp (Diagenode Bioruptor). Methylated DNA was then enriched using a Methylminer kit (Invitrogen) according to the manufactures protocol.Citation74 Briefly, the kit employs a biotin labeled methyl-binding domain from the human MBD2 protein to capture methylated DNA. When the captured DNA is eluted over an increasing stepwise salt gradient, DNA with fewer to higher methyl groups are separated. Following capture of sample DNA, salt washes were performed at 160 mM before collection of the 350 mM NaCl and 2000 mM NaCl elutions sequentially. Completely methylated and unmethylated control duplexes are included with the Methylminer kit and were used in parallel with each sample processed to validate the effectiveness and functionally of the enrichment. Both duplexes are 80 bp in length and contain either eight methylated or nine unmethylated CpGs. Sequencing libraries were constructed using the sequential 350 mM and 2000 mM sample elutions.
Sequencing library preparation
ChIP and Methylminer enriched sequencing libraries were prepared as per the ChIP-Seq library preparation kit protocol using single end adaptors (Illumina). The following changes were made to the protocol. “Variation to starting material”—sequencing libraries were constructed using 15 ng of sample DNA from Methylminer, ChIP bound, and ChIP input material. “Variation to 2% gel size selection of adaptor ligated material”—200–300 bp selection range was excised and collected for Methylminer material. For ChIP material, two size selections were taken, one selection range of 200–300 bp representing a largely mono-nucleosome fraction and another selection of 300–400 bp to represent material up to di-nucleosome in length. Gel size selection was based on the 1 Kb plus ladder (Invitrogen). PCR amplified libraries were diluted to 10 nM and stored at -20 °C in readiness for cluster generation and sequencing.
RNA-Seq
Total RNA was extracted from wild-type and MeCP2-null male mouse cerebellum using TRIZOL reagent (Invitrogen) and purified by column combined DNase digestion (Qiagen). Purified RNA was quantified by a qubit fluorometer (Invitrogen) and 5 µg per sample used in construction of sequencing libraries according to the mRNA-sequencing kit protocol (Illumina). PCR amplified libraries were diluted to 10 nM and stored at -20 °C in readiness for cluster generation and sequencing.
RIP-Seq (RNA-ChIP-Seq)
Purified RIP RNA (18 µL) was used in construction of sequencing libraries according to the NEBnext mRNA sample prep master mix set 1 protocol (New England Biolabs). PCR amplified libraries were diluted to 10 nM and stored at -20 °C in readiness for cluster generation and sequencing.
Massively parallel sequencing
Sequencing libraries were initially seeded to a flow cell and cluster amplified using the cluster kit version 4 protocol and cluster station system (Illumina). Massively parallel sequencing was performed using a Genome Analyzer IIx (Illumina), using the 36-cycle version 4 sequencing kit protocol (Illumina). Sequence and quality data were extracted from cluster cycle imaging using Pipeline v1.6 software (Illumina) and aligned to the mouse genome (July 2007 NCBI37/mm9 assembly) using the Burrows-Wheeler Aligner.Citation75 Sequencing data has been submitted to NCBI Gene Expression Omnibus (GEO) under the accession number GSE38324.
GEO data are currently not public and may be viewed using the reviewer link; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=hnwzxwkaukysaly&acc=GSE38324.
Bioinformatics tools
Genomic feature trends were analyzed with SeqMonk software package. DNA sequence of genomic regions and extended sequencing reads was extracted using Galaxy bioinformatics server tools. Sequence was quantified for CpG content or the regular expression describing CG-AT4 as CG (n ≤ 15) (A/T ≥ 4) where “n” represents number of random nucleotides, unless otherwise stated. Functional annotation of gene groups was performed using DAVID bioinformatics resources.Citation76,Citation77
RT-PCR
RNA was reverse transcribed using a high capacity cDNA reverse transcriptase Kit (Applied Biosystems). PCR amplification was performed using a 7500 Fast Real-Time PCR System (Applied Biosystems). Five pmoles of forward and reverse primer, 2 µL cDNA template and FAST SYBR® Green Master Mix (Roche) were mixed to a final volume of 13 µl with nuclease free water. RT-PCR cycle parameters were as follows- incubation at 95 °C for 10 min, followed by 50 cycles of - 95 °C for 3 s and 60 °C for 30 s. RIP enrichments were adjusted by input, and expressed relatively to GAPDH, while target gene expression levels were calculated as fold change normalized to GAPDH. Primers were as follows; GAPDH Forward 5′-TGAAGCAGGC ATCTGAGGG-3′, GAPDH Reverse 5′-CGAAGGTGGA AGAGTGGGAG -3′, RNCR3 e2–3 Forward 5′-GGGGTTCCGG GACAAAGCGT-3′, RNCR3 e2–3 Reverse 5′-CTCGCTTAGC TCAGAGGTCC GCA-3′, RNCR3 e2-I Forward 5′-ACGCTGCCCA ACAGTACCCG-3′, RNCR3 e2-I Reverse 5′-CACCCCACAG GGTTGGGGGA-3′, RNCR3 124a Forward 5′-AAGGCCTCTC TCTCCGTGTT-3′, RNCR3 124a Reverse 5′-CTCAGACAGC CCCATTCTTG-3′, RNCR3 e4 Forward 5′-CCTCCTTGGG GCTGTGGCTG-3′, RNCR3 e4 Reverse 5′-GAGCGATGGG GAGAGGCCCA-3′, MALAT1 mas2 Forward 5′-TGAGGCTGGG GAGTGTTCCA GT-3′, MALAT1 mas2 Reverse 5′-GACGGGGTTC AAGTCCCGCG-3′, MALAT1 e1 Forward 5′-GCCACACAGG AAGGCTCCGC-3′, MALAT1 e1 Reverse m 5′-GAGGGGTGAG GTGGGCGCTA-3′, MALAT1 mas1 Forward 5′-GGCTGGGGAG TGTTCCAGTG A-3′, MALAT1 mas1 Reverse 5′-TGGTGGCTGG CACTCCTGGT-3′.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Sources of Funding
The authors acknowledge grant and fellowship support from the National Health and Medical Research Council (NHMRC). Maxwell S was supported by a Postgraduate Research Scholarship and Pelka G by a Biomedical (Peter Doherty) Fellowship. Tam PPL is a Senior Principal Research Fellow and El-Osta A is a Senior Research Fellow of the NHMRC. Our work is supported in part by the Victorian Government’s Operational Infrastructure Support Program and Mr James Fairfax.
Acknowledgments
We sincerely thank Dr Said Sif for provision of Brg1 antibody and give special thanks to Dr Harikrishnan Kaipananickal, Antony Kaspi and Dr Mark Ziemann for their assistance with next generation sequencing and bioinformatic analysis, and Professor John Christodoulou for assistance with mouse models.
References
- Nan X, Campoy FJ, Bird A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell 1997; 88:471 - 81; http://dx.doi.org/10.1016/S0092-8674(00)81887-5; PMID: 9038338
- Meehan RR, Lewis JD, Bird AP. Characterization of MeCP2, a vertebrate DNA binding protein with affinity for methylated DNA. Nucleic Acids Res 1992; 20:5085 - 92; http://dx.doi.org/10.1093/nar/20.19.5085; PMID: 1408825
- Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet 1998; 19:187 - 91; http://dx.doi.org/10.1038/561; PMID: 9620779
- Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998; 393:386 - 9; http://dx.doi.org/10.1038/30764; PMID: 9620804
- Iguchi-Ariga SM, Schaffner W. CpG methylation of the cAMP-responsive enhancer/promoter sequence TGACGTCA abolishes specific factor binding as well as transcriptional activation. Genes Dev 1989; 3:612 - 9; http://dx.doi.org/10.1101/gad.3.5.612; PMID: 2545524
- Klose RJ, Sarraf SA, Schmiedeberg L, McDermott SM, Stancheva I, Bird AP. DNA binding selectivity of MeCP2 due to a requirement for A/T sequences adjacent to methyl-CpG. Mol Cell 2005; 19:667 - 78; http://dx.doi.org/10.1016/j.molcel.2005.07.021; PMID: 16137622
- Adams VH, McBryant SJ, Wade PA, Woodcock CL, Hansen JC. Intrinsic disorder and autonomous domain function in the multifunctional nuclear protein, MeCP2. J Biol Chem 2007; 282:15057 - 64; http://dx.doi.org/10.1074/jbc.M700855200; PMID: 17371874
- Ghosh RP, Nikitina T, Horowitz-Scherer RA, Gierasch LM, Uversky VN, Hite K, Hansen JC, Woodcock CL. Unique physical properties and interactions of the domains of methylated DNA binding protein 2. Biochemistry 2010; 49:4395 - 410; http://dx.doi.org/10.1021/bi9019753; PMID: 20405910
- Harikrishnan KN, Chow MZ, Baker EK, Pal S, Bassal S, Brasacchio D, Wang L, Craig JM, Jones PL, Sif S, et al. Brahma links the SWI/SNF chromatin-remodeling complex with MeCP2-dependent transcriptional silencing. Nat Genet 2005; 37:254 - 64; http://dx.doi.org/10.1038/ng1516; PMID: 15696166
- Nan X, Hou J, Maclean A, Nasir J, Lafuente MJ, Shu X, Kriaucionis S, Bird A. Interaction between chromatin proteins MECP2 and ATRX is disrupted by mutations that cause inherited mental retardation. Proc Natl Acad Sci U S A 2007; 104:2709 - 14; http://dx.doi.org/10.1073/pnas.0608056104; PMID: 17296936
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 2008; 320:1224 - 9; http://dx.doi.org/10.1126/science.1153252; PMID: 18511691
- Jeffery L, Nakielny S. Components of the DNA methylation system of chromatin control are RNA-binding proteins. J Biol Chem 2004; 279:49479 - 87; http://dx.doi.org/10.1074/jbc.M409070200; PMID: 15342650
- Buschdorf JP, Strätling WH. A WW domain binding region in methyl-CpG-binding protein MeCP2: impact on Rett syndrome. J Mol Med (Berl) 2004; 82:135 - 43; http://dx.doi.org/10.1007/s00109-003-0497-9; PMID: 14618241
- Young JI, Hong EP, Castle JC, Crespo-Barreto J, Bowman AB, Rose MF, Kang D, Richman R, Johnson JM, Berget S, et al. Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc Natl Acad Sci U S A 2005; 102:17551 - 8; http://dx.doi.org/10.1073/pnas.0507856102; PMID: 16251272
- Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron 2007; 56:422 - 37; http://dx.doi.org/10.1016/j.neuron.2007.10.001; PMID: 17988628
- Shahbazian MD, Antalffy B, Armstrong DL, Zoghbi HY. Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum Mol Genet 2002; 11:115 - 24; http://dx.doi.org/10.1093/hmg/11.2.115; PMID: 11809720
- Skene PJ, Illingworth RS, Webb S, Kerr AR, James KD, Turner DJ, Andrews R, Bird AP. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol Cell 2010; 37:457 - 68; http://dx.doi.org/10.1016/j.molcel.2010.01.030; PMID: 20188665
- Ghosh RP, Horowitz-Scherer RA, Nikitina T, Shlyakhtenko LS, Woodcock CL. MeCP2 binds cooperatively to its substrate and competes with histone H1 for chromatin binding sites. Mol Cell Biol 2010; 30:4656 - 70; http://dx.doi.org/10.1128/MCB.00379-10; PMID: 20679481
- Cohen S, Gabel HW, Hemberg M, Hutchinson AN, Sadacca LA, Ebert DH, Harmin DA, Greenberg RS, Verdine VK, Zhou Z, et al. Genome-wide activity-dependent MeCP2 phosphorylation regulates nervous system development and function. Neuron 2011; 72:72 - 85; http://dx.doi.org/10.1016/j.neuron.2011.08.022; PMID: 21982370
- Fraga MF, Ballestar E, Montoya G, Taysavang P, Wade PA, Esteller M. The affinity of different MBD proteins for a specific methylated locus depends on their intrinsic binding properties. Nucleic Acids Res 2003; 31:1765 - 74; http://dx.doi.org/10.1093/nar/gkg249; PMID: 12626718
- Bienvenu T, Chelly J. Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized. Nat Rev Genet 2006; 7:415 - 26; http://dx.doi.org/10.1038/nrg1878; PMID: 16708070
- Lintas C, Sacco R, Persico AM. Genome-wide expression studies in autism spectrum disorder, Rett syndrome, and Down syndrome. Neurobiol Dis 2012; 45:57 - 68; http://dx.doi.org/10.1016/j.nbd.2010.11.010; PMID: 21130877
- Leoncini S, De Felice C, Signorini C, Pecorelli A, Durand T, Valacchi G, Ciccoli L, Hayek J. Oxidative stress in Rett syndrome: natural history, genotype, and variants. Redox Rep 2011; 16:145 - 53; http://dx.doi.org/10.1179/1351000211Y.0000000004; PMID: 21888765
- Kelleher RJ 3rd, Bear MF. The autistic neuron: troubled translation?. Cell 2008; 135:401 - 6; http://dx.doi.org/10.1016/j.cell.2008.10.017; PMID: 18984149
- Kriaucionis S, Paterson A, Curtis J, Guy J, Macleod N, Bird A. Gene expression analysis exposes mitochondrial abnormalities in a mouse model of Rett syndrome. Mol Cell Biol 2006; 26:5033 - 42; http://dx.doi.org/10.1128/MCB.01665-05; PMID: 16782889
- Matzke MA, Birchler JA. RNAi-mediated pathways in the nucleus. Nat Rev Genet 2005; 6:24 - 35; http://dx.doi.org/10.1038/nrg1500; PMID: 15630419
- Zaratiegui M, Irvine DV, Martienssen RA. Noncoding RNAs and gene silencing. Cell 2007; 128:763 - 76; http://dx.doi.org/10.1016/j.cell.2007.02.016; PMID: 17320512
- Mattick JS, Amaral PP, Dinger ME, Mercer TR, Mehler MF. RNA regulation of epigenetic processes. Bioessays 2009; 31:51 - 9; http://dx.doi.org/10.1002/bies.080099; PMID: 19154003
- Kim DH, Villeneuve LM, Morris KV, Rossi JJ. Argonaute-1 directs siRNA-mediated transcriptional gene silencing in human cells. Nat Struct Mol Biol 2006; 13:793 - 7; http://dx.doi.org/10.1038/nsmb1142; PMID: 16936726
- Han J, Kim D, Morris KV. Promoter-associated RNA is required for RNA-directed transcriptional gene silencing in human cells. Proc Natl Acad Sci U S A 2007; 104:12422 - 7; http://dx.doi.org/10.1073/pnas.0701635104; PMID: 17640892
- Ting AH, Schuebel KE, Herman JG, Baylin SB. Short double-stranded RNA induces transcriptional gene silencing in human cancer cells in the absence of DNA methylation. Nat Genet 2005; 37:906 - 10; http://dx.doi.org/10.1038/ng1611; PMID: 16025112
- Jeon Y, Sarma K, Lee JT. New and Xisting regulatory mechanisms of X chromosome inactivation. Curr Opin Genet Dev 2012; 22:62 - 71; http://dx.doi.org/10.1016/j.gde.2012.02.007; PMID: 22424802
- Mercer TR, Dinger ME, Sunkin SM, Mehler MF, Mattick JS. Specific expression of long noncoding RNAs in the mouse brain. Proc Natl Acad Sci U S A 2008; 105:716 - 21; http://dx.doi.org/10.1073/pnas.0706729105; PMID: 18184812
- Li LC, Okino ST, Zhao H, Pookot D, Place RF, Urakami S, Enokida H, Dahiya R. Small dsRNAs induce transcriptional activation in human cells. Proc Natl Acad Sci U S A 2006; 103:17337 - 42; http://dx.doi.org/10.1073/pnas.0607015103; PMID: 17085592
- Politz JC, Zhang F, Pederson T. MicroRNA-206 colocalizes with ribosome-rich regions in both the nucleolus and cytoplasm of rat myogenic cells. Proc Natl Acad Sci U S A 2006; 103:18957 - 62; http://dx.doi.org/10.1073/pnas.0609466103; PMID: 17135348
- Hwang HW, Wentzel EA, Mendell JT. A hexanucleotide element directs microRNA nuclear import. Science 2007; 315:97 - 100; http://dx.doi.org/10.1126/science.1136235; PMID: 17204650
- Place RF, Li LC, Pookot D, Noonan EJ, Dahiya R. MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc Natl Acad Sci U S A 2008; 105:1608 - 13; http://dx.doi.org/10.1073/pnas.0707594105; PMID: 18227514
- Hamid QA, Thanumalayan S, Parnaik VK. An improved method to distinguish micrococcal nuclease sensitivity of chromatin. J Biochem Biophys Methods 1996; 33:59 - 64; http://dx.doi.org/10.1016/0165-022X(96)00014-0; PMID: 8905469
- Huang SY, Garrard WT. Electrophoretic analyses of nucleosomes and other protein-DNA complexes. Methods Enzymol 1989; 170:116 - 42; http://dx.doi.org/10.1016/0076-6879(89)70044-6; PMID: 2770536
- Singleton MK, Gonzales ML, Leung KN, Yasui DH, Schroeder DI, Dunaway K, LaSalle JM. MeCP2 is required for global heterochromatic and nucleolar changes during activity-dependent neuronal maturation. Neurobiol Dis 2011; 43:190 - 200; http://dx.doi.org/10.1016/j.nbd.2011.03.011; PMID: 21420494
- Nan X, Tate P, Li E, Bird A. DNA methylation specifies chromosomal localization of MeCP2. Mol Cell Biol 1996; 16:414 - 21; PMID: 8524323
- Lewis JD, Meehan RR, Henzel WJ, Maurer-Fogy I, Jeppesen P, Klein F, Bird A. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell 1992; 69:905 - 14; http://dx.doi.org/10.1016/0092-8674(92)90610-O; PMID: 1606614
- Choi JK. Contrasting chromatin organization of CpG islands and exons in the human genome. Genome Biol 2010; 11:R70; http://dx.doi.org/10.1186/gb-2010-11-7-r70; PMID: 20602769
- Andersson R, Enroth S, Rada-Iglesias A, Wadelius C, Komorowski J. Nucleosomes are well positioned in exons and carry characteristic histone modifications. Genome Res 2009; 19:1732 - 41; http://dx.doi.org/10.1101/gr.092353.109; PMID: 19687145
- Miranda TB, Jones PA. DNA methylation: the nuts and bolts of repression. J Cell Physiol 2007; 213:384 - 90; http://dx.doi.org/10.1002/jcp.21224; PMID: 17708532
- Yasui DH, Peddada S, Bieda MC, Vallero RO, Hogart A, Nagarajan RP, Thatcher KN, Farnham PJ, Lasalle JM. Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc Natl Acad Sci U S A 2007; 104:19416 - 21; http://dx.doi.org/10.1073/pnas.0707442104; PMID: 18042715
- Ricciardi S, Boggio EM, Grosso S, Lonetti G, Forlani G, Stefanelli G, Calcagno E, Morello N, Landsberger N, Biffo S, et al. Reduced AKT/mTOR signaling and protein synthesis dysregulation in a Rett syndrome animal model. Hum Mol Genet 2011; 20:1182 - 96; http://dx.doi.org/10.1093/hmg/ddq563; PMID: 21212100
- Guy J, Cheval H, Selfridge J, Bird A. The role of MeCP2 in the brain. Annu Rev Cell Dev Biol 2011; 27:631 - 52; http://dx.doi.org/10.1146/annurev-cellbio-092910-154121; PMID: 21721946
- Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet 2009; 10:155 - 9; http://dx.doi.org/10.1038/nrg2521; PMID: 19188922
- Kaludov NK, Wolffe AP. MeCP2 driven transcriptional repression in vitro: selectivity for methylated DNA, action at a distance and contacts with the basal transcription machinery. Nucleic Acids Res 2000; 28:1921 - 8; http://dx.doi.org/10.1093/nar/28.9.1921; PMID: 10756192
- El-Osta A, Kantharidis P, Zalcberg JR, Wolffe AP. Precipitous release of methyl-CpG binding protein 2 and histone deacetylase 1 from the methylated human multidrug resistance gene (MDR1) on activation. Mol Cell Biol 2002; 22:1844 - 57; http://dx.doi.org/10.1128/MCB.22.6.1844-1857.2002; PMID: 11865062
- Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, Greenberg ME. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 2003; 302:885 - 9; http://dx.doi.org/10.1126/science.1086446; PMID: 14593183
- Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 2003; 302:890 - 3; http://dx.doi.org/10.1126/science.1090842; PMID: 14593184
- McLachlan DR, Lewis PN, Lukiw WJ, Sima A, Bergeron C, De Boni U. Chromatin structure in dementia. Ann Neurol 1984; 15:329 - 34; http://dx.doi.org/10.1002/ana.410150405; PMID: 6742779
- Baker SA, Chen L, Wilkins AD, Yu P, Lichtarge O, Zoghbi HY. An AT-hook domain in MeCP2 determines the clinical course of Rett syndrome and related disorders. Cell 2013; 152:984 - 96; http://dx.doi.org/10.1016/j.cell.2013.01.038; PMID: 23452848
- Mellén M, Ayata P, Dewell S, Kriaucionis S, Heintz N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 2012; 151:1417 - 30; http://dx.doi.org/10.1016/j.cell.2012.11.022; PMID: 23260135
- Kass SU, Pruss D, Wolffe AP. How does DNA methylation repress transcription?. Trends Genet 1997; 13:444 - 9; http://dx.doi.org/10.1016/S0168-9525(97)01268-7; PMID: 9385841
- Bayles R, Harikrishnan KN, Lambert E, Baker EK, Agrotis A, Guo L, Jowett JB, Esler M, Lambert G, El-Osta A. Epigenetic modification of the norepinephrine transporter gene in postural tachycardia syndrome. Arterioscler Thromb Vasc Biol 2012; 32:1910 - 6; http://dx.doi.org/10.1161/ATVBAHA.111.244343; PMID: 22723437
- Harikrishnan KN, Bayles R, Ciccotosto GD, Maxwell S, Cappai R, Pelka GJ, Tam PP, Christodoulou J, El-Osta A. Alleviating transcriptional inhibition of the norepinephrine slc6a2 transporter gene in depolarized neurons. J Neurosci 2010; 30:1494 - 501; http://dx.doi.org/10.1523/JNEUROSCI.4675-09.2010; PMID: 20107077
- Ben-Shachar S, Chahrour M, Thaller C, Shaw CA, Zoghbi HY. Mouse models of MeCP2 disorders share gene expression changes in the cerebellum and hypothalamus. Hum Mol Genet 2009; 18:2431 - 42; http://dx.doi.org/10.1093/hmg/ddp181; PMID: 19369296
- Li Q, Lee JA, Black DL. Neuronal regulation of alternative pre-mRNA splicing. Nat Rev Neurosci 2007; 8:819 - 31; http://dx.doi.org/10.1038/nrn2237; PMID: 17895907
- Hartman TR, Qian S, Bolinger C, Fernandez S, Schoenberg DR, Boris-Lawrie K. RNA helicase A is necessary for translation of selected messenger RNAs. Nat Struct Mol Biol 2006; 13:509 - 16; http://dx.doi.org/10.1038/nsmb1092; PMID: 16680162
- Aratani S, Fujii R, Oishi T, Fujita H, Amano T, Ohshima T, Hagiwara M, Fukamizu A, Nakajima T. Dual roles of RNA helicase A in CREB-dependent transcription. Mol Cell Biol 2001; 21:4460 - 9; http://dx.doi.org/10.1128/MCB.21.14.4460-4469.2001; PMID: 11416126
- Abdelhaleem M, Maltais L, Wain H. The human DDX and DHX gene families of putative RNA helicases. Genomics 2003; 81:618 - 22; http://dx.doi.org/10.1016/S0888-7543(03)00049-1; PMID: 12782131
- Dardenne E, Pierredon S, Driouch K, Gratadou L, Lacroix-Triki M, Espinoza MP, Zonta E, Germann S, Mortada H, Villemin JP, et al. Splicing switch of an epigenetic regulator by RNA helicases promotes tumor-cell invasiveness. Nat Struct Mol Biol 2012; 19:1139 - 46; http://dx.doi.org/10.1038/nsmb.2390; PMID: 23022728
- Kuhn AN, Reichl EM, Brow DA. Distinct domains of splicing factor Prp8 mediate different aspects of spliceosome activation. Proc Natl Acad Sci U S A 2002; 99:9145 - 9; http://dx.doi.org/10.1073/pnas.102304299; PMID: 12087126
- Dellaire G, Makarov EM, Cowger JJ, Longman D, Sutherland HG, Lührmann R, Torchia J, Bickmore WA. Mammalian PRP4 kinase copurifies and interacts with components of both the U5 snRNP and the N-CoR deacetylase complexes. Mol Cell Biol 2002; 22:5141 - 56; http://dx.doi.org/10.1128/MCB.22.14.5141-5156.2002; PMID: 12077342
- Bernard D, Prasanth KV, Tripathi V, Colasse S, Nakamura T, Xuan Z, Zhang MQ, Sedel F, Jourdren L, Coulpier F, et al. A long nuclear-retained non-coding RNA regulates synaptogenesis by modulating gene expression. EMBO J 2010; 29:3082 - 93; http://dx.doi.org/10.1038/emboj.2010.199; PMID: 20729808
- Tripathi V, Ellis JD, Shen Z, Song DY, Pan Q, Watt AT, Freier SM, Bennett CF, Sharma A, Bubulya PA, et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell 2010; 39:925 - 38; http://dx.doi.org/10.1016/j.molcel.2010.08.011; PMID: 20797886
- Sanuki R, Onishi A, Koike C, Muramatsu R, Watanabe S, Muranishi Y, Irie S, Uneo S, Koyasu T, Matsui R, et al. miR-124a is required for hippocampal axogenesis and retinal cone survival through Lhx2 suppression. Nat Neurosci 2011; 14:1125 - 34; http://dx.doi.org/10.1038/nn.2897; PMID: 21857657
- Mercer TR, Qureshi IA, Gokhan S, Dinger ME, Li G, Mattick JS, Mehler MF. Long noncoding RNAs in neuronal-glial fate specification and oligodendrocyte lineage maturation. BMC Neurosci 2010; 11:14; http://dx.doi.org/10.1186/1471-2202-11-14; PMID: 20137068
- Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, Neul JL, Gong S, Lu HC, Heintz N, et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 2010; 468:263 - 9; http://dx.doi.org/10.1038/nature09582; PMID: 21068835
- Pelka GJ, Watson CM, Radziewic T, Hayward M, Lahooti H, Christodoulou J, Tam PP. Mecp2 deficiency is associated with learning and cognitive deficits and altered gene activity in the hippocampal region of mice. Brain 2006; 129:887 - 98; http://dx.doi.org/10.1093/brain/awl022; PMID: 16467389
- De Meyer T, Mampaey E, Vlemmix M, Denil S, Trooskens G, Renard JP, De Keulenaer S, Dehan P, Menschaert G, Van Criekinge W. Quality evaluation of methyl binding domain based kits for enrichment DNA-methylation sequencing. PLoS One 2013; 8:e59068; http://dx.doi.org/10.1371/journal.pone.0059068; PMID: 23554971
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25:1754 - 60; http://dx.doi.org/10.1093/bioinformatics/btp324; PMID: 19451168
- Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4:44 - 57; http://dx.doi.org/10.1038/nprot.2008.211; PMID: 19131956
- Huang W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009; 37:1 - 13; http://dx.doi.org/10.1093/nar/gkn923; PMID: 19033363