Abstract
Alternative splicing is a general mechanism for regulating gene expression that affects the RNA products of more than 90% of human genes. Not surprisingly, alternative splicing is observed among gene products of metazoan immune systems, which have evolved to efficiently recognize pathogens and discriminate between “self” and “non-self”, and thus need to be both diverse and flexible. In this review we focus on the specific interface between alternative splicing and autoimmune diseases, which result from a malfunctioning of the immune system and are characterized by the inappropriate reaction to self-antigens. Despite the widespread recognition of alternative splicing as one of the major regulators of gene expression, the connections between alternative splicing and autoimmunity have not been apparent. We summarize recent findings connecting splicing and autoimmune disease, and attempt to find common patterns of splicing regulation that may advance our understanding of autoimmune diseases and open new avenues for therapy.
Introduction
Alternative splicing is a highly elaborate process of gene regulation and a potent generator of protein diversity. Most metazoan protein-coding genes consist of short exons and much longer introns, which are retained and lost in mature mRNAs, respectively. For mRNAs to be transported to the cytoplasm where they can direct protein synthesis, introns need to be efficiently and accurately removed. This is achieved via the process of RNA splicing.Citation1 Considering that multiple mRNAs can be generated from a single pre-mRNA via different combinations of splicing patterns, and that splicing can be regulated in a variety of ways (tissue-specific, developmental stage-specific, stimulus-specific),Citation2 the resulting mRNA and protein diversity is truly enormous. According to some recent estimates,Citation3 up to 94% of all human genes undergo alternative splicing, which means that this mechanism of post-transcriptional regulation is critical for virtually all cellular processes.
Several layers of information, referred to as the “splicing code”, are included in the pre-mRNA sequence (reviewed in refs. Citation2–Citation5). These include the consensus 5′ and 3′ splice sites, a branch point sequence upstream of the 3′ splice site, and a wide variety of intronic and exonic splicing elements (see below). The basic splicing machinery, known as the spliceosome, recognizes and binds to these elements and executes a highly complicated task of correctly defining and joining appropriate exons together. Two cellular spliceosomes termed “major” and “minor”, work in parallel; here we will focus on the former. The major spliceosome, a multimegadalton ribonucleoprotein particle, consists of both RNAs (small nuclear RNAs, snRNAs: U1, U2, U4/U6 and U5) and a variety of protein factors (which together with the snRNAs form five small nuclear ribonucleoprotein complexes, or snRNPs).
The mechanisms of spliceosome assembly and action have been described in detail elsewhere (reviewed in refs. Citation6–Citation9). Briefly, the 5′ splice site is recognized first by the U1 snRNP, followed by the U2 snRNP binding to the branch point site, which results in the formation of the pre-splicing complex. Addition of the U4/U5/U6 tri-snRNP to the pre-splicing complex produces a fully assembled spliceosome, which undergoes conformational changes resulting in the first catalytic step of the splicing reaction, branch formation. After subsequent conformational changes and proper alignment of the two exons to be spliced, the second catalytic reaction, ligation of the exons, occurs. Importantly, spliceosome function is executed through dynamic RNA-RNA, RNA-protein and protein-protein interactions.Citation1,Citation10
Mammalian splice and branch point sites are short sequences, which do not contain all the information necessary to direct correct exon definition. This information is provided by additional splicing regulatory elements that exist within both exons and introns. Based on their mode of action, these additional elements can be divided into at least four groups: exonic or intronic splicing enhancers (ESEs or ISEs) activate splicing from within exons or introns, respectively, while exonic or intronic splicing silencers (ESSs or ISSs) suppress splicing.Citation2,Citation4,Citation11 Occasionally, the same element is able to activate and suppress splicing of two mutually exclusive exons (reviewed in ref. Citation12). Most of the aforementioned cis-acting regulatory elements function through the binding of different trans-acting protein factors such as heterogeneous nuclear ribonucleoproteins, hnRNPs and SR (serine/argininerich) proteins. The mechanism by which many of these cis-elements and trans-acting factors regulate splicing is, however, still not completely understood.
The layers of regulation described above control both constitutive and alternative splicing of exons. Alternative splicing, which leads to the generation of multiple different transcripts from the same primary template, proceeds through several different pathways (): (a) use of alternative 5′ splice sites; (b) use of alternative 3′ splice sites; (c) intron retention; (d) a choice to include or skip an exon, and (e) a choice between two mutually exclusive exons.Citation5,Citation13 Together with other events that lead to transcript diversity (e.g., alternative promoter usage and/or alternative polyadenylation)Citation3 the five processes described in result in protein isoforms that can possess diverse or even opposite functions.
The extraordinarily high degree of proteome diversity generated by alternative splicing, as well as the number of genes that undergo this processing, implies that alternative splicing regulates virtually every aspect of cellular metabolism, and, as a consequence, every system in the human body. The immune system is no exception. Evolved to protect the body from remarkably diverse pathogens and to discriminate between “self” and “nonself”, the immune system requires diversity of gene expression, and the contribution of alternative splicing to this diversity has only recently begun to be appreciated.
Many genes in the immune system have been shown to undergo alternative splicing (e.g., HLA, TCRζ-chain, a variety of cytokine and cytokine receptor transcripts; for an excellent review on the importance of alternative splicing in the immune system, see ref. Citation13). Nevertheless, our knowledge of the global involvement of alternative splicing in different aspects of immunity is still limited. Microarrays and other high-throughput technologies provide an invaluable tool to study global gene expression on a genome-wide scale. When applied to studying genes involved in T- and B-cell activation, microarray analysis using Affymetrix Human Exon 1.0 ST arrays reveals three distinct classes of genes that change as a function of immune activation in both T and B cells: (1) differentially expressed and alternatively spliced; (2) constitutively expressed and alternatively spliced, and (3) differentially expressed without alternative splicing.Citation14 Interestingly, of all the differentially expressed genes, about 44% are alternatively spliced at each time point of T-cell activation, while of all the constitutively expressed genes, 50–70% are alternatively spliced. This finding shows that there is potential for a significant increase in transcript diversity due to alternative splicing of constitutively expressed genes upon T-cell activation. These genome-wide approaches should reveal the global impact of alternative splicing on the immune system.
Alternative splicing and autoimmune diseases.
As noted above, alternative splicing is critical for normal functioning of virtually every system in the human body. At the same time, it is also implicated in the development of various diseases, and many disease-associated missense mutations mediate their deleterious effects by altering the splicing pattern of the gene.Citation2,Citation15 Autoimmunity, or the inappropriate reaction to self-antigens, underlies a number of diseases, some organ-specific and some systemic, and represents an exciting, yet complex, field of study. Many (if not all) autoimmune diseases are thought to be complex, with both environmental and multiple genetic factors determining pathology. An understanding of the importance of alternative splicing in the development of autoimmune diseases is now just starting to emerge. As is the case for any other disease in which aberrant splicing plays a role, autoimmune diseases can result from mutations in both cis-acting regulatory elements and trans-acting factors. As reviewed in ref. Citation4, mutations in any of these elements can: (1) cause disease directly; (2) modify its severity; (3) confer susceptibility. Moreover, processing and presentation of antigens, both of “self” and “foreign” origin, can be influenced by alternative splicing (see below). Overall, deciphering the connections between alternative splicing and autoimmunity holds promise for a better understanding of both processes.
While a number of genes have been found to be associated with autoimmune diseases, we will focus this review on multiple sclerosis, a major immune-mediated neurological disorder, and discuss the data that point to a role of alternative splicing in this disease. We will then briefly outline how other autoimmune diseases may be affected by alternative splicing.
Multiple Sclerosis
Multiple sclerosis (MS) is a major neurodegenerative disorder involving the central nervous system (CNS). Disease features include penetration of lymphocytes into the CNS, induction of local inflammation and immune responses, demyelination and axon damage.Citation16 MS preferentially affects young adults and shows a strong gender preference with females being affected 2–3 times more frequently than males.Citation17 The course of the disease varies greatly among individual patients, ranging from a mild, or even asymptomatic, disease to severe disability. MS has been divided into four clinical subtypes as a means of predicting prognosis and response to therapy: relapsing remitting (85% of diagnosed MS cases), primary progressive (10%), progressive relapsing (5%) and secondary progressive (develops in 50–80% of relapsing remitting patients).Citation16 The prevalence is highest among individuals of Caucasian origin with more than 400,000 individuals affected in the US and more than 800,000 in Europe. MS is a disease with a strong genetic component; however, environmental factors (e.g., viral infection) have also been proposed to play a role.Citation16
Multiple family-based and case-control studies have been performed on MS populations to identify disease susceptibility genes. However, for almost forty years the only genetic locus consistently linked to the disease has been the human leukocyte antigen locus, or HLA. This changed in 2007 when a SNP within the gene encoding the interleukin 7 receptor alpha chain (IL7R) was shown to be significantly associated with susceptibility to MS (see below). Importantly, alternative splicing of IL7R transcripts was affected by this MS-associated allele. We will review the data supporting this finding and then focus our attention on other mRNA transcripts that are potentially regulated by alternative splicing in MS.
IL7R and MS
The receptor for IL7 consists of two chains: the IL7R chain (CD127), which provides ligand specificity, and the common cytokine receptor γc-chain (CD132). IL7R is expressed almost exclusively on cells of the lymphoid lineage, and it is crucial for their survival and proliferation. IL7R expression is tightly regulated, strictly based on whether or not these cells need to receive survival, proliferation, or, in some cases, differentiation signals. IL7R is expressed at the double negative (CD4-CD8-) T-cell progenitor stage, absent at the double positive (CD4+CD8+) stage, and then re-expressed at the single positive (CD4+ or CD8+) stage. In addition to T-cell development in the thymus, naïve and memory T cells in the periphery require signaling through IL7R for their survival. The significance of this tightly regulated expression of IL7R has been noted and is reviewed elsewhere.Citation18
Association of IL7R with susceptibility to MS has been suggested by a number of studies, however, this association was unambiguously established and replicated by three partially independent research groups.Citation19–Citation21 A functional, non-synonymous single-nucleotide polymorphism (SNP) rs6897932 (T→C, T244I) within exon 6 of IL7R shows the strongest association with MS among all three studies. The result for rs6897932 is due to over-transmission of the “C” risk allele to offspring affected with MS and is independent of the known HLA effect.Citation19
IL7R pre-mRNA consists of eight exons, with exon 6 coding for the transmembrane domain. Two isoforms of IL7R have been identified based on alternative splicing of exon 6: a membrane-bound isoform (rIL7R), in which exon 6 is included, and a soluble isoform (sIL7R), in which exon 6 is skipped ().Citation22,Citation23 Importantly, both isoforms are capable of ligand (IL7) binding.
To test whether alternative splicing of exon 6 was differentially affected in transcripts from the “T” or the MS-associated “C” alleles, Gregory et al.Citation19 analyzed exon 6 inclusion both in vitro and in vivo. For the in vitro analysis, minigenes containing either “T” or “C” alleles of rs6897932, as well as parts of flanking introns, were created. When transfected into a variety of cell lines, transcripts from the minigenes containing the MS-associated “C” allele show an approximately two-fold increase in exon 6 skipping when compared with transcripts containing the “T” allele. Based on additional mutagenesis analysis (transversions to either “G” or “A” at the SNP position, as well as substitutions around the SNP), the authors concluded that the disease-associated “C” allele affects exon 6 alternative splicing by augmenting the action of an exonic splicing silencer (ESS).
The in vitro results were supported by at least two lines of evidence in vivo: first, when peripheral blood mononuclear cells (PBMC) from healthy controls are analyzed by quantitative real-time PCR for allele-specific IL7R expression, a significantly lower expression of the exon 6–7 amplicon is observed for carriers of the “C” allele.Citation19 Second, semi-quantitative RT-PCR analysis of PBMC from MS patients who are homozygous for the “C” allele showed a 4–5-fold increase in exon 6 skipping compared to patients who are homozygous for the “T” allele (Gregory et al., unpublished results). Additionally, Lundmark et al.Citation20 reported that expression of IL7R mRNA was elevated in the cerebrospinal fluid of individuals with MS in comparison with individuals with other neurological diseases. This suggests that changes in IL7R expression have pathophysiological significance, although no distinction between the two IL7R isoforms was made. Lastly, McKay et al.Citation24 established that expression of sIL7R mRNA was significantly elevated in whole blood samples from patients with two types of MS (primary progressive and relapsing remitting), and that increased expression of sIL7R correlated with a particular IL7R haplotype that was more common in the primary progressive MS patients. Taken together, these data suggest that: (1) there is a decrease in the ratio of transmembrane to soluble isoforms of IL7R in MS, and (2) aberrant alternative splicing of IL7R exon 6 causes this change in isoform ratio.
Clearly, the causality of IL7R in MS pathogenesis has not been formally proven, and the exact role of the increased production of sIL7R in MS has not been unraveled. One can envision, however, a number of plausible explanations that could connect aberrant splicing of exon 6, increased sIL7R production and MS. As discussed earlier, developing T cells in the thymus as well as naïve and memory T cells in the periphery require signaling through IL7R for survival and proliferation. As reviewed in ref. Citation18, IL7R ligand, IL7, is expressed constitutively; however, its amount is thought to be very low and just sufficient to maintain a finite number of T cells. Therefore, lower expression of the transmembrane IL7R, due to increased skipping of exon 6, could lower IL7-mediated signaling below a critical threshold and compromise survival and proliferation of T cells (in this delicately balanced system, even a small decrease in rIL7R would be sufficient to generate an effect). Additionally, sequestration of the ligand by the increased amounts of the sIL7R isoform could deprive T cells of their survival and proliferative signals. It must be noted, however, that the connection between this antagonistic mode of action for sIL7R and the development of MS, as proposed here, remains unproven.
Developing a hypothesis in which aberrant signaling through rIL7R, and, thus, compromised T-cell survival and proliferation, contribute to the etiology of MS is challenging for two reasons: (1) other unknown environmental and genetic factors may contribute to the development of the disease, and (2) the possible commensurate decrease in T-cell populations is contrary to a disease model in which there is acute T cell proliferation and inflammatory response that results in axon demyelination. Two studies that identified an increase in CD8+ T cells from PBMC in MS patientsCitation25,Citation26 also identified a relative decrease in CD8+ T cells in the CNS, in line with the assumption that a decrease in T-cell populations is associated with the disease.Citation26 It is also possible that lower expression of rIL7R by CD8+ T cells in MS patients renders these cells more susceptible to viral attack. Indeed, various viruses have been implicated in the development of MS,Citation27,Citation28 and in chronic viral infections a phenomenon known as CD8+ T cell exhaustion, which is characterized by poor function and viability of memory T cells, as well as low levels of rIL7R expression by these cells, has been described.Citation18,Citation29,Citation30 Thus, it is possible that in individuals who genetically express lower levels of rIL7R, CD8+ T-cell exhaustion occurs in response to chronic viral infection. Viral infection can subsequently induce autoimmunity, resulting in demyelination and axon damage.
CTLA-4: A General Marker of Autoimmunity?
Cytotoxic T-lymphocyte antigen 4 (CTLA-4) is another candidate for association with MS. Several genetically associated polymorphisms have been identified within the CTLA-4 gene, and multiple attempts have been made to establish their role in susceptibility to MS and other autoimmune diseases (reviewed in ref. Citation31). Yet, reports on the association of CTLA-4 with MS have been inconsistent.Citation32–Citation41 However confusing the association of CTLA-4 with MS, it is worth considering this gene as a potential contributor to the disease.
CTLA-4 is critical for negative regulation of T-cell proliferation. By binding to its ligands (CD80 and CD86) on antigen-presenting cells, CTLA-4 counteracts the activity of the T-cell costimulatory molecule CD28, which binds to the same ligands to activate T-cell proliferation. Negative regulation of T-cell proliferation by CTLA-4 results in the induction of T-cell anergy, and, eventually, peripheral tolerance.Citation42 Together with the data from experiments in CTLA-4-knockout mice, which develop lymphoproliferative disorders characterized by infiltration of lymphocytes into multiple organs and tissue destruction,Citation43 and CTLA-4-knockdown animals, which rapidly progress to autoimmune diabetes,Citation44 these results suggest a role of CTLA-4 in the development of autoimmunity.
Two isoforms of CTLA-4 are produced by alternative splicing in human cells: a “full-length” transmembrane isoform (flCTLA-4), in which exons 1 through 4 are included, and a soluble isoform (sCTLA-4), in which exon 3 is skipped.Citation45,Citation46 Both CTLA-4 isoforms are capable of binding CD80 and CD86. Interestingly, production of these isoforms seems to be regulated by T cell activation: resting T cells express sCTLA-4, but downregulate its expression upon activation, whereas expression of flCTLA-4 rapidly increases upon activation and remains constant until the end of the immune response.Citation13,Citation47,Citation48 Considering that regulation of CTLA-4 expression is critical for providing proper downregulation signals for activated T cells, and that availability of the transmembrane isoform could affect signaling through CTLA-4, it is attractive to propose that aberrant differential expression of CTLA-4 isoforms, either due to alternative splicing, or other mechanisms, could contribute to autoimmune diseases.
Is alternative splicing of CTLA-4 important for the development of other autoimmune diseases? Abundant data implicate CTLA-4 in a number of autoimmune diseases (autoimmune hypothyroidism, Graves' disease, type I diabetes,Citation31 and Myasthenia gravis,Citation49 to name a few), and therefore, CTLA-4 has been proposed as a “general susceptibility gene to autoimmunity”.Citation33 Interestingly, an abnormal splicing spectrum of CTLA-4 isoforms has been detected in patients with Myasthenia gravis,Citation49 and recent data suggest that when the levels of sCTLA-4 are reduced by RNAi, the potency of regulatory T cells is significantly reduced.Citation50 These two lines of evidence underscore the importance of CTLA-4 alternative splicing in the development of autoimmune diseases.
CD45, An Intriguing Example with an Uncertain Association
CD45, a prototypic receptor tyrosine phosphatase, is essential for activation and development of T cells.Citation51 Alternative splicing of exons 4–6 within the CD45 extracellular domain results in the production of isoforms CD45RABC (exons 4–6 included), CD45RAB (exons 4 and 5 included), CD45RBC (exons 5 and 6 included), CD45RB (exon 5 included), and CD45RO (exons 4–6 skipped). Naïve T cells express longer isoforms at higher levels, while activated and memory T cells express shorter isoforms (CD45RO).Citation13,Citation52 In resting T cells, CD45 phosphatase activity is needed to maintain a signal-competent pool of protein kinases of the Src family, which initiates a signal transduction cascade and induces T-cell activation upon antigen encounter. Importantly, this phosphatase activity can be regulated by isoform-differential homodimerization: the alternatively spliced isoforms differentially homodimerize in primary T cells, with the smallest isoform (CD45RO) being able to dimerize more efficiently and rapidly than the larger isoforms.Citation52CD45 dimers have significantly reduced phosphatase activity, which implies that this splicing switch is critical for preventing prolonged TCR signaling and undesirable tissue injury.Citation52
Several SNPs that affect alternative splicing of exon 4 in CD45 have been identified.Citation53,Citation54 Some of these SNPs have been proposed to be associated with MS.Citation53 For example, the SNP 77C→G in exon 4 results in increased production of the longer isoforms. Considering that the longer isoforms are associated with more efficient TCR signaling, failure to attenuate prolonged TCR activation could contribute to the inflammatory response resulting in axon demyelination in MS. In contrast, the nonsynonymous SNP 138G→A in exon 6 results in increased production of the shorter isoform CD45RO,Citation55,Citation56 with the “G” allele having a significant protective effect in Graves' disease, another autoimmune disorder.Citation55 Other studies, however, have argued against a significant association between CD45 and MS.Citation57–Citation62 Alternative splicing of CD45 is one of the best-characterized splicing events in the immune system, both at the level of cis-acting regulatory elements,Citation63 and trans-acting protein factors.Citation64–Citation67 Pending resolution of the conflicting reports on association with MS, these studies could prove valuable for the understanding of alternative splicing in this disorder.
Other MS Susceptibility Genes with Alternative Splicing Connections
OAS1.
Haplotype “GA” of the 2′,5′-oligoadenylate synthetase 1 (OAS1) gene, defined by the “G” allele of a SNP at the 3′ splice site of exon 7 and the “A” allele of a SNP in exon 3, has been linked to increased OAS1 enzyme activity as well as MS and type 1 diabetes susceptibility.Citation68–Citation71 Importantly, different alternatively spliced mRNA isoforms are produced depending on which allele is present at the 3′ splice site of exon 7.Citation69 However, functional data establishing the role of these alternative splicing products and their corresponding protein counterparts in susceptibility to MS are lacking. It is tempting to speculate that involvement of OAS1 in the pathogenesis of this disease reflects possible viral etiology. OAS1 is one of the critical components of the interferon antiviral response; it is activated in the presence of double-stranded viral RNAs and catalyzes polymerization of ATP into 2′,5′-linked oligoadenylates, which bind to and activate the latent ribonuclease, RNase L, resulting in degradation of viral RNAs.Citation69 It is necessary to establish the role(s) that different alternatively spliced OAS1 isoforms play in both defense against viruses and MS pathogenesis. Caution, however, needs to be taken when assessing the contribution of individual OAS1 SNPs to the development of MS and other autoimmune diseases, due to the long-range linkage disequilibrium between OAS1 polymorphisms,Citation68,Citation71 as well as recently reported inconsistencies in association of OAS1 with type 1 diabetes.Citation72,Citation73
KLC1.
One report suggests a possible involvement of a particular variant of the kinesin light-chain 1 (KLC1) gene in protection against MS.Citation74 Kinesin is involved in trafficking of the myelin synthesis apparatusCitation74,Citation75 (hence a direct connection to MS), and a protective SNP within intron 13 of the KLC1 gene has been hypothesized to affect splicing. To our knowledge, no data supporting this hypothesis exist; however, it is of interest that multiple splicing isoforms of KLC1 have been identified.Citation76 If the SNP in intron 13 is, in fact, involved in protection against MS, it might shed light on the role of the alternatively spliced kinesin isoforms in MS.
Alternative Splicing of Candidate Autoantigens, Splicing Factors and More
Potential autoantigens.
Autoantigen isoforms generated by alternative splicing could potentially exhibit different immunogenic properties and are extremely attractive candidates for detailed splicing analysis. Interestingly, NgCitation77,Citation78 and colleagues applied bioinformatics analyses to determine the extent of alternative splicing within 45 randomly selected human autoantigens and randomly selected, non-autoantigenic genes. Based on these analyses, alternative splicing occurs in transcripts of 100% of autoantigens, and only in about 42% of randomly selected genes. Although these results need to be re-interpreted in light of recent reports suggesting that almost every gene in the human genome can be alternatively spliced,Citation3 they suggest that autoantigen transcripts are more prone to splicing regulation. Moreover, splicing by the minor spliceosome (involving U12-type introns) is predicted to occur more frequently in autoantigen transcripts than in transcripts from non-autoantigens.Citation77 The authorsCitation77,Citation78 suggest that alternative splicing can modulate imunogenicity of different spliced forms by: (1) creating novel antigenic epitopes; (2) altering surface accessibility; (3) enabling expression by tissues not normally expressing autoantigens, and (4) altering patterns of posttranslational modifications. Thus, alternative splicing may be responsible, at least in part, for the generation of untolerized epitopes, and, as a result, autoimmunity.
Several candidate autoantigens have been identified in MS, namely, myelin basic protein (MBP), myelin oligodendrocyte glycoprotein (MOG), and myelin proteolipid protein (PLP), as well as others. Transcription from a large Golli (for “gene expressed in the oligodendrocyte lineage”)-MBP transcription unit gives rise to Golli-MBP and classic MBP pre-mRNAs, both of which are alternatively spliced, producing different protein isoforms. Processing of these protein isoforms by the enzymes of the matrix metalloproteinase (MMP) family can generate similar sets of immunogenic peptides.Citation79–Citation81 Moreover, while expression of classic MBP isoforms is restricted to myelin-forming cells in the CNS, the Golli-MBP alternatively spliced transcripts are expressed in the thymus, spleen and lymph nodes, as well as on the cells of the myeloid lineage, providing possible MS autoantigens outside of the CNS. Therefore, it is plausible that these MBP-containing Golli-MBP isoforms, processed by MMPs into immunogenic peptides, are recognized by autoreactive T cells which, if they cross the blood-brain barrier, would recognize neuronal MBP as an antigen.
Myelin oligodendrocyte glycoprotein (MOG) is a minor integral membrane protein found on the outermost surface of the myelin sheath. Complex alternative splicing of the MOG pre-mRNA gives rise to up to seventeen different isoforms (both trans-membrane and secreted).Citation82 Investigation of the six major mRNA isoforms shows that they: (1) localize differentially (cell surface vs. endoplasmic reticulum vs. secreted) and (2) are translated.Citation83,Citation84 Although analysis of the MS brain tissue samples did not reveal any disease-specific differences in the MOG isoform patterns,Citation83 it is still possible that MOG isoforms generated by alternative splicing could be involved in MS pathogenesis. Secreted forms of MOG can have a protective effect if they interact with anti-MOG antibodies and thus prevent demyelination, while CNS-specific forms can induce an autoimmune response when recognized by specific anti-MOG antibodies upon the death of oligodendrocytes and subsequent exposure of novel MOG antigens.Citation83
Finally, use of alternative 5′ splice sites within exon 3 of the gene coding for the main component of CNS myelin, myelin proteolipid protein (PLP), gives rise to two alternatively spliced isoforms: the longer PLP isoform and the shorter DM20 isoform (reviewed in refs. Citation81, Citation85–Citation89, and also see ). DM20 lacks exon 3B and codes for a protein product that is 35 amino acids shorter than the PLP isoform. Interestingly, despite its major role in the CNS, PLP is also expressed outside of the CNS, most importantly, in the thymus, with intrathymic expression largely restricted to the short DM20 splice variant. In a study using a model of MS known as experimental autoimmune encephalomyelitis (EAE), Klein et al.Citation90 show that the thymic expression of DM20 is sufficient to confer T-cell tolerance to all epitopes of the PLP protein in EAE-resistant mice. In contrast, one of the PLP epitopes in EAE-susceptible mice is encoded by the CNS-specific exon 3B of PLP, which is skipped in the DM20 splice variant. Thus, EAE-susceptible mice are prone to developing the disease due to the lack of tolerance to this particular epitope of the PLP protein. As the expression of PLP in the human thymus is also restricted to the DM20 isoform,Citation90 an “uncensored” repertoire of T cells that escape central tolerance and are autoreactive to the exon 3B-encoded epitope, exists. Once damage to myelin by as-yet-undefined triggers has occurred, and full-length PLP has been presented, these specific autoreactive T cells may be activated and gain access to the CNS, where they can initiate an inflammatory response and thus contribute to MS.Citation90
PLP/DM20 alternative splicing has been characterized in detail.Citation86–Citation89 Several cis-acting regulatory elements, including G-rich sequences, have been found in exon 3B. Two of these G-rich elements, named G1 and M2, bind hnRNPs H and F, which recruit U1 snRNP to the DM20 5′ splice site and regulate the PLP/DM20 ratio.Citation89 These mechanistic data, although not yet connected to the development of MS or changes in T-cell tolerance, are invaluable for understanding PLP/DM20 splicing regulation and its potential involvement in MS pathogenesis.
Viral splicing?
The involvement of viral infection in MS pathogenesis has been debated for quite a while and has been reviewed elsewhere.Citation28,Citation91 The human endogenous retrovirus HERV-H has been proposed to be associated with MS, and interestingly, one study reported that a combination of two HERV-H splice variants, the so called “envelope” (env) and “protease-envelope” (protease-env) splice variants, can be found in 40% of MS patients but only in 10% of controls, which, the authors speculated, might be a sign of the activated state of the virus and may even lead to increased production of the HERV-H env protein.Citation92 In another study, a possibility that alternative splicing plays a role in generating different isoforms for yet another MS-associated human endogenous retrovirus, HERV-W, has been suggested.Citation93 Involvement of alternatively spliced viral transcripts in MS pathogenesis is an untested, albeit intriguing, hypothesis.
Alternative splicing and demyelination.
As described earlier, regulated expression of the myelin protein components is important for establishing proper myelination patterns and can potentially be affected in MS. A classic demyelination model in the mouse is the quakingviable (qkv) mutation. The mutation results from the deletion of an upstream regulatory sequence in the qkI gene, leading to reduced transcription of the QKI protein, a single hnRNP K homology (KH) RNA-binding domain protein.Citation94–Citation96 Qkv mice have a twitching phenotype and show severe demyelination of the CNS, with reduced levels of QKI correlating with the severity of demyelination. Interestingly, different QKI protein isoforms are produced by alternative splicing in a developmentally-regulated manner.Citation94–Citation96 Several lines of evidence have established that some of these isoforms serve as splicing factors to regulate expression of myelin structural genes (MBP, PLP and myelin-associated glycoprotein, MAG). For example, isoform QKI-5 binds to an intronic splicing silencer (ISS) within intron 12 of the MAG pre-mRNA and represses exon 12 inclusion in wild-type mice.Citation96 In the qkv mice, however, where less QKI protein is present, more isoform with exon 12 included is produced.Citation96 QKI-5 also regulates splicing of PLP (by switching the splicing towards the DM20 isoform) and MBP (by preferentially including exon 6 and excluding exon 5) pre-mRNAs, implying that aberrant splicing regulation is caused by reduced levels of QKI protein in the mutant mice and lies at the heart of the demyelination phenotype.
Although no data regarding the involvement of the human homolog of QKI in demyelination in MS are available, the quaking model presents an interesting example of an important event in MS pathology that is modulated by alternative splicing. It would be interesting to establish whether similar misregulation of QKI homologs is observed in MS.
Other Autoimmune Diseases
Alternative splicing affects more than 90% of the genes in the human genome, so it would not be surprising if most of the genes involved in autoimmunity would have at least one alternatively spliced isoform. Both mechanistic and functional data on alternatively spliced genes in autoimmunity are still lacking (as most of the reports simply propose alternative splicing involvement without follow-up confirmation). Regardless of this, a trend is apparent: most of the genes involved in autoimmunity are alternatively spliced, and a vast area of research aimed at the mechanistic determination of splicing pathways and the role of alternative splicing in developing autoimmunity is now open. Some of the genes proposed to be alternatively spliced in autoimmune diseases are listed in . A thorough description of all of these genes and the alternative processing of their transcripts is beyond the scope of this review; thus, we chose to focus on some selected examples.
Myasthenia gravis.
Myasthenia gravis (MG) is an autoimmune disease characterized by impaired transmission across the neuromuscular junctions and consequent muscle weakness.Citation97 MG is mediated by autoantibodies against the nicotinic acetylcholine receptor (nAchR). Inhibitors of acetylcholinesterase (AChE), an enzyme that degrades acetylcholine, have been used widely for the treatment of MG, but an unexpected side effect caused by alternative splicing of AChE transcripts calls their use into question. Two isoforms of AChE, the major (synaptic) isoform, AChE-S, and a normally rare readthrough isoform, AChE-R, are non-selectively blocked by these inhibitors.Citation98 These isoforms are differentially induced under stress, and destruction of nAChR by MG antibodies, followed by cholinergic imbalance, is a stress condition that results in a splicing switch towards production of the readthrough isoform. Unexpectedly, treatment with non-selective AChE blockers contributes to elevated AChE-R production. Because AChE-R is soluble and secreted, and capable of non-synaptic hydrolysis of acetylcholine, its elevated expression exacerbates muscle weakness. Interestingly, an antisense oligonucleotide that selectively binds to this particular spliced isoform has been tested in the rat model of MG, and its administration results in improved survival, neuromuscular strength and clinical status in the animals tested.Citation98 This anti-sense oligonucleotide is being evaluated as a potential anti-MG drug in clinical trials.Citation99
An aforementioned alternative splicing event among CTLA-4 transcripts has also been proposed to be associated with MG (ref. Citation100, and also see the above discussion of the role of CTLA-4 in multiple sclerosis and general autoimmunity).
Ulcerative colitis.
Ulcerative colitis (UC), a subgroup of inflammatory bowel disease, is a chronic debilitating intestinal illness. A three-marker SNP haplotype within the receptor protein-tyrosine phosphatase sigma (PTPσ) gene has been found to be significantly associated with UC.Citation101 The presence of the haplotype results in a novel splicing event, skipping of exons 8 and 9, which leads to the deletion of the entire third immunoglobulin domain in the PTPσ ectodomain. Although the functions of the deleted domain, as well as the consequences of its deletion, are still unclear, it is possible that the new isoform possesses either different dimerization or ligand-binding abilities. PTPσ is thought to maintain its substrates, E-cadherin and β-catenin, in a dephosphorylated state, which is necessary for preventing the disassembly of the adherens junction, and aberrant alternative splicing associated with the disease-susceptible haplotype is likely to interfere with this or other PTPσ functions. Importantly, PTPσ knockout mice develop mild colitis, and are susceptible to developing severe colitis when challenged with specific colitis inducers,Citation101 supporting the role of PTPσ in the development of UC.
Kawasaki disease.
A pediatric systemic vasculitis of unknown etiology, Kawasaki disease has been proposed to involve T-cell activation and infiltration of coronary artery walls.Citation102 A functional SNP itpkc_3 (G→C) in intron 1 of the inositol 1,4,5-triphosphate 3-kinase C (ITPKC) gene is significantly associated with disease susceptibility and formation of coronary artery aneurysms. The presence of the “C” risk allele results in reduced ITPKC splicing when minigene constructs are employed in in vitro splicing assays.Citation102 Since it is thought that ITPKC serves as a negative regulator of T-cell activation, it seems likely that a reduction in splicing will lead to lower levels of functional ITPKC, an increase in T-cell activation, and subsequent inflammation.
At the nucleotide level, the disease-associated “C” allele disrupts a “GGG” run, and is located right next to the 5′ splice site of intron 1. Poly-G runs constitute splicing enhancer elements and are important regulators of splicing, which are known to serve as binding sites for hnRNP H; recent work has shown that the activity of the poly-G runs depends on the strength of the adjacent 5′ splice site (it is about 4-fold higher when adjacent to an intermediate-strength 5′ splice site as compared to a weak 5′ splice site, and about 1.3-fold higher when compared to a strong 5′ splice site).Citation103 The 5′ splice site of ITPKC intron 1 conforms to the consensus sequence. Therefore, the activity of hnRNP H would depend on both of these elements. It is likely that the disease-associated “C” allele disrupts the poly-G enhancer element, interferes with hnRNP H binding, and leads to reduced ITPKC splicing. Although direct regulation of this splicing event by binding of hnRNP H has yet to be proven experimentally, this is an excellent example of how recent advances in bioinformatics analyses of alternative splicing, combined with genetic data, reveal the mechanistic aspects of splicing regulation.
Systemic lupus erythematosus.
Systemic lupus erythematosus (SLE) is a prototypic systemic autoimmune disease.Citation104 SLE is predominant among young women and is characterized by the presence of autoantibodies against a variety of antigens. Unknown etiology, coupled with the ability of SLE to involve any or all organ systems in its pathogenesis, makes this disease particularly interesting and important to study.
Multiple SLE susceptibility genes have been identified, and despite some inconsistency in the results, it is very likely that many genes that are crucial for SLE pathogenesis have already been discovered.Citation105 Interestingly, some of them are proposed to affect splicing (). For example, three SNPs in the BANK1 (B-cell scaffold protein with ankyrin repeats) gene together confer susceptibility to SLE. One of these SNPs, located at the branch-point site within intron 1, affects the relative splicing efficiency and upregulates production of a novel BANK1 isoform that lacks exon 2.Citation106 As a result, this BANK1 isoform lacks a putative inositol 1,4,5-triphosphate receptor (IP3R)-binding domain and is proposed to act in a dominant-negative or dose-dependent manner to attenuate BANK1-mediated signaling.Citation106 The precise role of the attenuated signaling of BANK1 in SLE is still unclear, as is the role of BANK1 in B cell receptor-mediated signaling, but considering the crucial role of B cells in the pathogenesis of SLE, it is likely that aberrant BANK1 functioning is involved in pathogenesis.
Leukocyte immunoglobulin-like receptor A2 (LILRA2) is a stimulatory receptor that is highly expressed on monocytes, macrophages, myeloid denritic cells and a subset of natural killer cells. A SNP G→A that disrupts a 3′ splice site of intron 6 in the LILRA2 pre-mRNA, has been associated with SLE in a Japanese population.Citation107 The presence of the SLE-associated allele activates a cryptic 3′ splice site 9 nucleotides downstream of the original 3′ splice site. This leads to production of a novel protein isoform that lacks three amino acids in the linker region and is expressed on the surface of monocytes. Since the ligands for LILRA2 are still undetermined, it is hard to predict how the three amino acid deletion affects ligand binding; however, the authors speculate that either signaling through Toll-like receptors or regulation of a specific subpopulation of T helper cells could be affected by this deletion. Both hypotheses still remain to be tested, and an independent confirmation of the association of LILRA2 with SLE in a different population is needed.
Regulation of the T-cell receptor ζ chain (TCRζ) pre-mRNA by alternative splicing is yet another example of the potential importance of alternative splicing in SLE.Citation108–Citation110 Several alternatively spliced isoforms of TCRζ have been reported, including a form lacking exon 7, as well as an isoform in which the 3′-untranslated region is alternatively spliced. Production of these two isoforms leads to reduced expression of both the TCR/CD3 complex and the ζ protein itself on the cell surface, as well as reduced production of interleukin 2 (IL2) upon stimulation. Moreover, both isoforms are less stable than the full-length transcripts, and their more rapid degradation contributes to diminished expression of the TCR. protein. The multi-subunit T-cell receptor (TCR)/CD3 complex uses TCRζ chains for signal transduction, so abnormalities in the expression of the TCRζ chain can lead to T-cell dysfunction, loss of tolerance and the development of autoimmunity.
Other genes regulated at the level of alternative splicing have been associated with SLE and other autoimmune diseases (). Discussing them in detail goes beyond the scope of this review, but from the number of studies that have revealed a possible connection between alternative splicing and SLE pathogenesis, one can conclude that alternative splicing regulation is important for conferring the risk to SLE and contributing to its pathogenesis.
Concluding Remarks
Cellular gene expression is regulated at many levels, all of which contribute to the final amount and diversity of gene products. Given that nearly all gene products undergo alternative splicing, the extent to which expression of genes is regulated by this processing is truly extraordinary. Moreover, many disease-associated missense mutations exert their deleterious effect by altering the splicing pattern of the gene.
The immune system, which recognizes and fights a seemingly endless variety of “foreigners”, but should not react towards the equally diverse “self” components, thrives on diversity and flexibility, with alternative splicing being one of the major contributors. At the same time, when the immune system malfunctions, for example, when it inappropriately reacts towards “self” determinants, alternative splicing is likely to be involved. Until recently this connection was not immediately apparent. This was due to a number of reasons: first, in many cases, splicing of a particular disease-associated gene has been proposed, but not confirmed, at the RNA or protein level; second, even if the presence of alternatively spliced isoforms at the RNA level was confirmed, data connecting the corresponding protein isoforms with pathogenesis were lacking. Moreover, in the majority of cases, mechanistic studies to decipher how splicing of each particular susceptibility gene works, e.g., by characterizing its cis-acting regulatory elements and trans-acting factors, have not been performed. Lastly, inconsistency between studies trying to ascertain genetic effects that, although real, are small, adds to the confusion and sometimes makes even thoroughly performed mechanistic studies inapplicable to the analysis of splicing in autoimmunity.
Regardless of these difficulties, we have summarized evidence that indicates an important role of alternative splicing in autoimmunity and highlighted the following common trends: first, as the case of IL7R exemplifies, alternative splicing can produce soluble counterparts to transmembrane cytokine receptors, and these soluble isoforms can be associated with disease. Other examples include the involvement of the soluble receptor for interleukin 6 (sIL6R) in the pathophysiology of inflammatory bowel disease and rheumatoid arthritis and possible involvement of a novel isoform of the soluble receptor for tumor necrosis factor α [DS-TNFR2(Δ7,8)] in rheumatoid arthritis.Citation111–Citation113 Second, since all autoimmune diseases share one unique feature—production of autoantigens—generation of different autoantigen isoforms by alternative splicing can potentially lead to creation of new immunogenic epitopes. Although attractive, this hypothesis needs thorough experimental testing, which may prove difficult due to the mere fact that in many cases the causative autoantigens have not been identified.
Overall, revealing the connections between alternative splicing and the pathogenesis of autoimmune diseases is a complex, yet exciting, task. Thorough studies on both the mechanism of splicing regulation as well as the role that alternatively spliced isoforms play in etiology or pathogenesis, are needed in each case. This will not only reveal how alternative splicing of particular genes operates, but will also shed light onto some general mechanisms of alternative splicing involvement in the pathogenesis of autoimmunity.
Abbreviations
| CNS | = | central nervous system |
| EAE | = | experimental autoimmune encephalomyelitis |
| ESE | = | exonic splicing enhancer |
| ESS | = | exonic splicing silencer |
| HLA | = | human leukocyte antigen |
| hnRNP | = | heterogeneous nuclear ribonucleoprotein |
| IL7R | = | interleukin 7 receptor α chain |
| ISE | = | intronic splicing enhancer |
| ISS | = | intronic splicing silencer |
| MS | = | multiple sclerosis |
| PBMC | = | peripheral blood mononuclear cell |
| pre-mRNA | = | pre-messenger RNA |
| RNAi | = | RNA interference |
| RT-PCR | = | reverse transcription-polymerase chain reaction |
| SNP | = | single-nucleotide polymorphism |
| SR | = | serine/arginine-rich protein |
| TCRζ | = | T-cell receptor ζ chain |
Figures and Tables
Figure 1 Alternative pre-mRNA splicing. (A) alternative use of 5′ splice sites; (B) alternative use of 3′ splice sites; (C) intron retention; (D) alternative use of a cassette exon; (E) alternative use of two mutually exclusive exons. For explanations, see text.
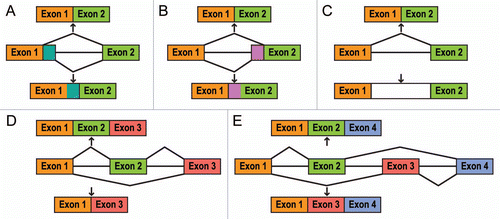
Figure 2 Schematic representation of IL7R pre-mRNA splicing. Genomic and pre-mRNA structure of the IL7R gene is shown (top), with SNPs selected for genotyping in reference Citation19, listed above. The MS-associated SNP, rs6897932, is highlighted. Alternative splicing of the IL7R pre-mRNA leads to the production of two isoforms: a membrane-bound (rIL7R, exon 6 included) or soluble (sIL7R, exon 6 skipped). The transmembrane isoform of the receptor consists of the extracellular (EX), transmembrane (TM) and cytoplasmic (CYTO) domains. Skipping of exon 6 leads to a translation reading frame shift and translation termination due to a premature stop codon. Adapted in part from reference Citation23.
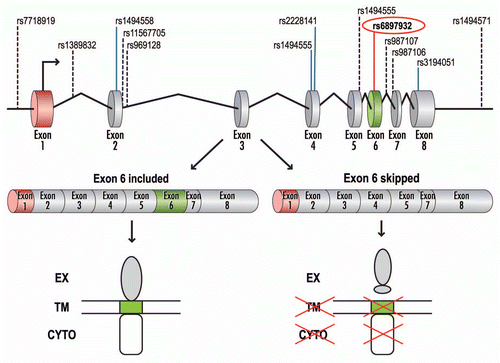
Table 1 Alternatively spliced genes that are implicated in autoimmune diseases
Acknowledgement
The authors thank Dr. Shelton Bradrick for critical reading of this document and many useful comments. The authors acknowledge support from grants NIH R01 CA127727 (M.G.B.) and NIH R01 NS060925-01 (S.G.).
References
- Tycowski KT, Kolev NG, Conrad NK, Fok V, Steitz JA. Gesteland RF, Cech TR, Atkins JF. The ever-growing world of small nuclear ribonucleoproteins. The RNA World 2006; Third edition NY CSHL Press 329 - 336
- Cooper TA, Wan L, Dreyfuss G. RNA and disease. Cell 2009; 136:777 - 793
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, et al. Alternative isoform regulation in human tissue transcriptomes. Nature 2008; 456:470 - 476
- Wang G-S, Cooper TA. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev 2007; 8:749 - 761
- Goldstrohm AC, Greenleaf AL, Garcia-Blanco MA. Co-transcriptional splicing of pre-messenger RNAs: considerations for the mechanism of alternative splicing. Gene 2001; 277:31 - 47
- Wahl MC, Will CL, Lührmann R. The spliceosome: design principles of a dynamic RNP machine. Cell 2009; 136:701 - 718
- Staley JP, Woolford JL Jr. Assembly of ribosomes and spliceosomes: complex ribonucleoprotein machines. Curr Opin Cell Biol 2009; 21:109 - 118
- Smith DJ, Query CC, Konarska MM. Nought may endure but mutability: spliceosome dynamics and the regulation of splicing. Mol Cell 2008; 30:657 - 666
- Jurica MS. Detailed close-ups and the big picture of spliceosomes. Curr Opin Struct Biol 2008; 18:315 - 320
- Patel AA, Steitz JA. Splicing double: insights from the second spliceosome. Nat Rev Mol Cell Biol 2003; 4:960 - 970
- Garcia-Blanco MA, Baraniak AP, Lasda EL. Alternative splicing in disease and therapy. Nat Biotechnol 2004; 22:535 - 546
- Carstens RP, McKeehan WL, Garcia-Blanco MA. An intronic sequence element mediates both activation and repression of rat fibroblast growth factor receptor 2 pre-mRNA splicing. Mol Cell Biol 1998; 18:2205 - 2217
- Lynch KW. Consequences of regulated pre-mRNA splicing in the immune system. Nat Rev 2004; 4:931 - 940
- Grigoryev YA, Kurian SM, Nakorchevskiy AA, Burke JP, Campbell D, Head SR, et al. Genome-wide analysis of immune activation in human T and B cells reveals distinct classes of alternatively spliced genes. PLoS ONE 2009; 4:e7906
- Lopez-Bigas N, Audit B, Ouzounis C, Parra G, Guigó R. Are splicing mutations the most frequent cause of hereditary disease?. FEBS Lett 2005; 579:1900 - 1903
- Coyle PK. Lahita RG. Multiple sclerosis. Textbook of the autoimmune diseases 2000; Lippincott Williams & Wilkins 595 - 609
- Duquette P, Pleines J, Girard M, Charest L, Senecal-Quevillon M, Masse C. The increased susceptibility of women to multiple sclerosis. Can J Neurol Sci 1992; 19:466 - 471
- Mazzucchelli R, Durum SK. Interleukin-7 receptor expression: intelligent design. Nat Rev Immunol 2007; 7:144 - 153
- Gregory SG, Schmidt S, Seth P, Oksenberg JR, Hart J, Prokop A, et al. Interleukin 7 receptor a chain (IL7R) shows allelic and functional association with multiple sclerosis. Nat Genetics 2007; 39:1083 - 1091
- Lundmark F, Duvefelt K, Iacobaeus E, Kockum I, Wallström E, Khademi M, et al. Variation in interleukin 7 receptor a chain (IL7R) influences risk of multiple sclerosis. Nat Genetics 2007; 39:1108 - 1113
- The International Multiple Sclerosis Genetics Consortium. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med 2007; 357:851 - 862
- Goodwin RG, Friend D, Ziegler SF, Jerzy R, Falk BA, Gimpel S, et al. Cloning of the human and murine interleukin-7 receptors: demonstration of a soluble form and homology to a new receptor superfamily. Cell 1990; 60:941 - 951
- Korte A, Köchling J, Badiali L, Eckert C, Andreae J, Geilen W, et al. Expression analysis and characterization of alternatively spliced transcripts of human IL7Rα chain encoding two truncated receptor proteins in relapsed childhood ALL. Cytokine 2000; 12:1597 - 1608
- McKay FC, Swain LI, Schibeci SD, Rubio JP, Kilpatrick TJ, Heard RN, et al. Haplotypes of the interleukin7 receptor alpha gene are correlated with altered expression in whole blood cells in multiple sclerosis. Genes Immun 2008; 9:1 - 6
- Liu GZ, Fang LB, Hjelmstrom P, Gao XG. Increased CD8+ central memory T cells in patients with multiple sclerosis. Mult Scler 2007; 13:149 - 155
- Haegele KF, Stueckle CA, Malin JP, Sindern E. Increase of CD8+ T-effector memory cells in peripheral blood of patients with relapsing-remitting multiple sclerosis compared to healthy controls. J Neuroimmunol 2007; 183:168 - 174
- Lunemann JD, Munz C. Epstein-Barr virus and multiple sclerosis. Curr Neurol Neurosci Rep 2007; 7:253 - 258
- Grigoriadis N, Hadjigeorgiuo GM. Virus-mediated autoimmunity in multiple sclerosis. J Autoimmune Dis 2006; 3:1 - 8
- Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 2006; 443:350 - 354
- Mueller SN, Ahmed R. High antigen levels are the cause of T cell exhaustion during chronic viral infection. Proc Natl Acad Sci USA 2009; 106:8623 - 8628
- Ueda H, Howson JMM, Esposito L, Heward J, Snook H, Chamberlain G, et al. Association of the T-cell regulatory gene CTLA-4 with susceptibility to autoimmune disease. Nature 2003; 423:506 - 511
- Ligers A, Xu C, Saarinen S, Hillert J, Olerup O. The CTLA-4 gene is associated with multiple sclerosis. J Neuroimmunol 1999; 97:182 - 190
- Kristiansen OP, Larsen ZM, Pociot F. CTLA-4 in autoimmune diseases—a general susceptibility gene to autoimmunity?. Genes and Immunity 2000; 1:170 - 184
- Ligers A, Teleshova N, Masterman T, Huang W-X, Hillert J. CTLA-4 gene expression is influenced by promoter and exon 1 polymorphisms. Genes Immun 2001; 2:145 - 152
- Kantarci OH, Hebrink DD, Achenbach SJ, Atkinson EJ, Waliszewska A, Buckle G, et al. CTLA-4 is associated with susceptibility to multiple sclerosis. J Neuroimmunol 2003; 134:133 - 141
- Van Veen T, Crusius J, Bart A, van Winsen L, Xia B, Barkhof F, Salvador Pena A, et al. CTLA-4 and CD28 gene polymorphisms in susceptibility, clinical course and progression of multiple sclerosis. J Neuroimmunol 2003; 140:188 - 193
- Anjos SM, Shao W, Marchand L, Polychronakos C. Allelic effects on gene regulation at the autoimmunity- predisposing CTLA-4 locus: a re-evaluation of the 3′ + 6230G>A polymorphism. Genes Immun 2005; 6:305 - 311
- Roxburgh RH, Sawcer S, Maranian M, Seaman S, Hensiek A, Yeo T, et al. No evidence of a significant role for CTLA-4 in multiple sclerosis. J Neuroimmunol 2006; 171:193 - 197
- Bagos PG, Karnaouri AC, Nikolopoulos GK, Hamodrakas SJ. No evidence for association of CTLA-4 gene polymorphisms with the risk of developing multiple sclerosis: a meta-analysis. Multiple Sclerosis 2007; 13:156 - 168
- Palacios R, Comas D, Elorza J, Villoslada P. Genomic regulation of CTLA-4 and multiple sclerosis. J Neuroimmunol 2008; 203:108 - 115
- Karabon L, Kosmaczewska A, Bilinska M, Pawlak E, Ciszak L, Jedynak A, et al. The CTLA-4 gene polymorphisms are associated with CTLA-4 protein expression levels in multiple sclerosis patients and with susceptibility to disease. Immunology 2009; 128:787 - 796
- Carreno BM, Bennett F, Chau TA, Ling V, Luxenberg D, Jussif J, et al. CTLA-4 (CD152) can inhibit T cell activation by two different mechanisms depending on its level of cell surface expression. J Immunol 2000; 165:1352 - 1356
- Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 1995; 270:985 - 988
- Chen Z, Stockton J, Mathis D, Benoist C. Modeling CTLA-4-linked autoimmunity with RNA interference in mice. Proc Natl Acad Sci USA 2006; 103:16400 - 16405
- Oaks MK, Hallett KM, Penwell RT, Stauber EC, Warren SJ, Tector AJ. A native soluble form of CTLA-4. Cell Immunol 2000; 201:144 - 153
- Pawlak E, Kochanowska IE, Frydecka I, Kielbinski M, Potoszek S, Bilinska M. The soluble CTLA-4 receptor: a new marker in autoimmune diseases. Arch Immunol Ther Exp 2005; 53:336 - 341
- Perkins D, Wang Z, Donovan C, He H, Mark D, Guan G, et al. Regulation of CTLA-4 expression during T cell activation. J Immunol 1996; 156:4154 - 4159
- Magistrelli G, Jeannin P, Herbault N, Benoit de Coignac A, Gauchat JF, Bonnefoy JY, et al. A soluble form of CTLA-4 generated by alternative splicing is expressed by nonstimulated human T cells. Eur J Immunol 1999; 29:3596 - 3602
- Gu M, Kakoulidou M, Giscombe R, Pirskanen R, Lefvert AK, Klareskog L, et al. Identification of CTLA-4 isoforms produced by alternative splicing and their association with Myasthenia gravis. Clin Immunol 2008; 128:374 - 381
- Kissler S, Fischer K, Zheng P, Rainbow D, Wicker L. The soluble CTLA-4 splice variant affects the function of CD4+CD25+ regulatory T cells. J Immunol 2009; 182:49 - 10
- Holmes N. A splicing switch for T cells. Science 2008; 321:646 - 647
- Hermiston ML, Xu Z, Weiss A. CD45: a critical regulator of signaling thresholds in immune cells. Annu Rev Immunol 2003; 21:107 - 137
- Jacobsen M, Schweer D, Ziegler A, Gaber R, Schock S, Schwinzer R, et al. A point mutation in PTPRC is associated with the development of multiple sclerosis. Nat Genetics 2000; 26:495 - 498
- Jacobsen M, Hoffmann S, Cepok S, Stei S, Ziegler A, Sommer N, et al. A novel mutation in PTPRC interferes with splicing and alters the structure of the human CD45 molecule. Immunogenetics 2002; 54:158 - 163
- Boxall S, Stanton T, Hirai K, Ward V, Yasui T, Tahara H, et al. Disease associations and altered immune function in CD45 138G variant carriers. Hum Mol Gen 2004; 13:2377 - 2384
- Ward V, Hennig BJ, Hirai K, Tahara H, Tamori A, Dawes R, et al. Geographical distribution and disease associations of the CD45 exon 6 138G variant. Immunogenetics 2006; 58:235 - 239
- Gomez-Lira M, Liguori M, Magnani C, Bonamini D, Salviati A, Leone M, et al. CD45 and multiple sclerosis: the exon 4 C77G polymorphism (additional studies and meta-analysis) and new markers. J Neuroimmunol 2003; 140:216 - 221
- Vorechovsky I, Kralovicova J, Tchilian E, Masterman T, Zhang Z, Ferry B, et al. Does 77C→G in PTPRC modify autoimmune disorders linked to the major histocompatibility complex?. Nat Genetics 2001; 29:22 - 23
- Barcellos LF, Caillier S, Dragone L, Elder M, Vittinghoff E, Bucher P, et al. PTPRC (CD45) is not associated with the development of multiple sclerosis in US patients. Nat Genet 2001; 29:23 - 24
- Miterski B, Sindern E, Haupts M, Schimrigk S, Epplen JT. PTPRC (CD45) is not associated with multiple sclerosis in a large cohort of German patients. BMC Med Gen 2002; 3:1 - 3
- Cocco E, Murru MR, Melis C, Schirru L, Solla E, Lai M, et al. PTPRC (CD45) C77G mutation does not contribute to multiple sclerosis susceptibility in Sardinian patients. J Neurol 2004; 251:1085 - 1088
- Tchilian EZ, Dawes R, Ramaley PA, Whitworth JA, Yuldasheva N, Spencer Wells R, et al. A CD45 polymorphism associated with abnormal splicing is absent in African populations. Immunogenetics 2002; 53:9803
- Lynch KW, Weiss A. A CD45 polymorphism associated with multiple sclerosis disrupts an exonic splicing silencer. J Biol Chem 2001; 26:24341 - 24347
- Topp JD, Jackson J, Melton AA, Lynch KW. A cell-based screen for splicing regulators identifies hnRNP LL as a distinct signal-induced repressor of CD45 variable exon 4. RNA 2008; 14:2038 - 2049
- Oberdoerffer S, Ferreira Moita L, Neems D, Freitas RP, Hacohen N, Rao A. Regulation of CD45 alternative splicing by heterogeneous ribonucleoprotein, hnRN-PLL. Science 2008; 321:686 - 691
- Wu Z, Jia X, de la Cruz L, Su X-C, Marzolf B, Troisch P, et al. Memory T cell RNA rearrangement programmed by heterogeneous nuclear ribonucleoprotein hnRNPLL. Immunity 2008; 29:863 - 875
- Wang H-Y, Xu X, Ding J-H, Bermingham JR Jr, Fu X-D. SC35 plays a role in T cell development and alternative splicing of CD45. Mol Cell 2001; 7:331 - 342
- Fedetz M, Matesanz F, Caro-Maldonado A, Fernandez O, Tamayo JA, Guerrero M, et al. OAS1 gene haplo-type confers susceptibility to multiple sclerosis. Tissue Antigens 2006; 68:446 - 449
- Bonnevie-Nielsen V, Leigh Field L, Lu S, Zheng D-J, Li M, Martensen PM, et al. Variation in antiviral 2′,5′-oligoadenylate synthetase (2′5′AS) enzyme activity is controlled by a single-nucleotide polymorphism at a splice-acceptor site in the OAS1 gene. Am J Hum Genet 2005; 76:623 - 633
- Leigh Field L, Bonnevie-Nielsen V, Pociot F, Lu S, Nielsen TB, Beck-Nielsen H. OAS1 splice site polymorphism controlling antiviral enzyme activity influences susceptibility to type 1 diabetes. Diabetes 2005; 54:1588 - 1591
- Tessier M-C, Qu H-Q, Frechette R, Bacot F, Grabs R, Taback SP, et al. Type 1 diabetes and the OAS gene cluster: association with splicing polymorphism or haplotype?. J Med Genet 2006; 43:129 - 132
- Smyth DJ, Cooper JD, Lowe CE, Nutland S, Walker NM, Clayton DG, et al. No evidence for association of OAS1 with type 1 diabetes in unaffected siblings or type 1 diabetic cases. Diabetes 2006; 55:1525 - 1528
- Qu HQ. Polychronakos C, Type 1 Diabetes Genetics Consortium. Reassessment of the type 1 diabetes association of the OAS1 locus. Genes Immun 2009; 10:69 - 73
- Szolnoki Z, Kondacs A, Mandi Y, Somogyvari F. A cytoskeleton motor protein genetic variant may exert a protective effect on the occurrence of multiple sclerosis: the janus face of the kinesin light-chain 1 56836CC genetic variant. Neuromol Med 2007; 9:335 - 339
- Carson JH, Worboys K, Ainger K, Barbarese E. Translocation of myelin basic protein mRNA in oligodendrocytes requires microtubules and kinesin. Cell Motil Cytoskeleton 1997; 38:318 - 328
- McCart AE, Mahony D, Rothnagel JA. Alternatively spliced products of the human kinesin light chain 1 (KNS2) gene. Traffic 2003; 4:576 - 580
- Ng B, Yang F, Huston DP, Yan Y, Yang Y, et al. Increased noncanonical splicing of autoantigen transcripts provides the structural basis for expression of untolerized epitopes. J Allergy Clin Immunol 2004; 114:1463 - 1470
- Yang D, Chen IH, Xiong Z, Yan Y, Wang H, Yang X-F. Model of stimulation-responsive splicing and strategies in identification of immunogenic isoforms of tumor antigens and autoantigens. Clin Immunol 2006; 121:121 - 133
- Shiryaev SA, Savinov AY, Cieplak P, Ratnikov BI, Motamedchaboki K, Smith JW, et al. Matrix metalloproteinase proteolysis of the myelin basic protein isoforms is a source of immunogenic peptides in auto-immune multiple sclerosis. PLoS ONE 2009; 4:4952
- Pribyl TM, Campagnoni CW, Kampf K, Kashima T, Handley VW, McMahon J, et al. The human myelin basic protein gene is included within a 179-kilobase transcription unit: expression in the immune and central nervous systems. PNAS 1993; 90:10695 - 10699
- Pribyl TM, Campagnoni C, Kampf K, Handley VS, Campagnoni AT. The major myelin protein genes are expressed in the human thymus. J Neurosci Res 1996; 45:812 - 819
- Delarasse C, Della Gaspera B, Lu CW, Lachapelle F, Gelot A, Rodriguez D, et al. Complex alternative splicing of the myelin oligodendrocyte glycoprotein gene is unique to human and non-human primates. J Neurochem 2006; 98:1707 - 1717
- Boyle LH, Traherne JA, Plotnek G, Ward R, Trowsdale J. Splice variation in the cytoplasmic domains of myelin oligodendrocyte glycoprotein affects its cellular localization and transport. J Neurochem 2007; 102:18536 - 18562
- Allamargot C, Gardinier MV. Alternative isoforms of myelin/oligodendrocyte glycoprotein with variable cytoplasmic domains are expressed in human brain. J Neurochem 2007; 101:298 - 312
- Pribyl TM, Campagnoni CW, Kampf K, Kashima T, Handley VW, McMahon J, Campagnoni AT. Expression of the myelin proteolipid protein gene in the human fetal thymus. J Neuroimmunol 1996; 67:125 - 130
- Wang E, Huang Z, Hobson GM, Dimova N, Sperle K, McCullough A, et al. PLP1 alternative splicing in differentiating oligodendrocytes: characterization of an exonic splicing enhancer. J Cell Biochem 2005; 97:999 - 1016
- Wang E, Dimova N, Cambi F. PLP/DM20 ratio is regulated by hnRNPH and F and a novel G-rich enhancer in oligodendrocytes. NAR 2007; 35:4164 - 4178
- Wang E, Dimova N, Sperle K, Huang Z, Lock L, McCullough MC, et al. Deletion of a splicing enhancer disrupts PLP1/DM20 ratio and myelin stability. Exp Neurol 2008; 214:322 - 330
- Wang E, Cambi F. Heterogenous nuclear ribonucleoproteins H and F regulate the proteolipid protein/DM20 ratio by recruiting U1 small nuclear ribonucleoprotein through a complex array of G runs. J Biol Chem 2009; 284:11194 - 11204
- Klein L, Klugmann M, Nave K-A, Tuohy VK, Kyewski B. Shaping of the autoreactive T-cell repertoire by a splice variant of self protein expressed in thymic epithelial cells. Nat Med 2000; 6:56 - 61
- Perron H, Lang A. The human endogenous retrovirus link between genes and environment in multiple sclerosis and in multifactorial diseases associating neuroinflammation. Clin Rev Allergy Immunol 2009; http://dx.doi.org/10.1007/s12016-009-8170-x
- Christensen T, Sorensen PD, Hansen HJ, Moller-Larsen A. Antibodies against a human endogenous retrovirus and the preponderance of env splice variants in multiple sclerosis patients. Mult Scler 2003; 9:6 - 15
- Blond J-L, Beseme F, Duret L, Bouton O, Bedin F, Perron H, et al. Molecular characterization and placental expression of HERV-W, a new human endogenous retrovirus family. J Virol 1999; 73:1175 - 1185
- Zhao L, Tian D, Xia M, Macklin WB, Feng Y. Rescuing qkv dysmyelination by a single isoform of the selective RNA-binding protein QKI. J Neurosci 2006; 26:11278 - 11286
- Ryder SP, Williamson JR. Specificity of the STAR/GSG domain protein Qk1: implications for the regulation of myelination. RNA 2004; 10:1449 - 1458
- Wu JI, Reed RB, Grabowski PJ, Artzt K. Function of quaking in myelination: regulation of alternative splicing. Proc Natl Acad Sci USA 2002; 99:4233 - 4238
- Rider LG, Targoff IN. Lahita RG. Muscle diseases. Textbook of the autoimmune diseases 2000; Lippincott Williams & Wilkins 462 - 467
- Brenner T, Hamra-Amitay J, Evron T, Boneva N, Seidman S, Soreq H. The role of readthrough acetylcholinesterase in the pathophysiology of myasthenia gravis. FASEB J 2003; 17:214 - 222
- Sussman JD, Argov Z, McKee D, Hazum E, Brawer S, Soreq H. Antisense treatment for Myasthenia gravis. Experience with Monarsen. Ann NY Acad Sci 2008; 1132:283 - 290
- Gu M, Kakoulidou M, Giscombe R, Pirskanen R, Lefvert AK, Klareskog L, et al. Identification of CTLA-4 isoforms produced by alternative splicing and their association with myasthenia gravis. Clinical Immunol 2008; 128:374 - 381
- Muise AM, Walters T, Wine E, Griffiths AM, Turner D, Duerr RH, et al. Protein-tyrosine phosphatase sigma is associated with ulcerative colitis. Curr Biol 2007; 17:1212 - 1218
- Onouchi Y, Gunji T, Burns JC, Shimizu C, Newburger JW, Yashiro M, et al. ITPKC functional polymorphism associated with Kawasaki disease susceptibility and formation of coronary artery aneurysms. Nat Genet 2008; 40:35 - 42
- Xiao X, Wang Z, Jang M, Nutiu R, Wang ET, Burge CB. Splice site strength-dependent activity and genetic buffering by poly-G runs. Nat Struct Mol Biol 2009; 16:1094 - 1100
- Lahita RG. Lahita RG. Systemic Lupus Erythematosus. Textbook of the autoimmune diseases 2000; Lippincott Williams & Wilkins 537 - 547
- Sestak AL, Nath SK, Sawalha AH, Harley JB. Current status of lupus genetics. Arthritis Res Ther 2007; 9:210
- Kozyrev SV, Abelson A-K, Wojcik J, Zaghlool A, Prasad Linga Reddy MV, Sanchez E, et al. Functional variants in the B-cell gene BANK1 are associated with systemic lupus erythematosus. Nat Genet 2008; 40:211 - 216
- Mamegano K, Kuroki K, Miyashita R, Kusaoi M, Kobayashi S, Matsuta K, et al. Association of LILRA2 (ILT1, LIR7) splice site polymorphism with systemic lupus erythematosus and microscopic polyangiitis. Genes Immun 2008; 9:214 - 223
- Tsuzaka K, Setoyama Y, Yoshimoto K, Shiraishi K, Suzuki K, Abe T, et al. A splice variant of the TCRζ mRNA lacking exon 7 leads to the downregulation of TCRζ, the TCR/CD3 complex, and IL-2 production in systemic lupus erythematosus T cells. J Immunol 2005; 174:3518 - 3525
- Tsuzaka K, Fukuhara I, Setoyama Y, Yoshimoto K, Suzuki K, Abe T, et al. TCRζ mRNA with an alternatively spliced 3′-untranslated region detected in systemic lupus erythematosus patients leads to the downregulation of TCRζ and TCR/CD3 complex. J Immunol 2003; 171:2496 - 2503
- Tsuzaka K, Nozaki K, Kumazawa C, Shiraishi K, Setoyama Y, Yoshimoto K, et al. TCRζ mRNA splice variant forms observed in the peripheral blood T cells from systemic lupus erythematosus patients. Springer Semin Immun 2006; 28:185 - 193
- Rose-John S, Waetzig GH, Scheller J, Grotzinger J, Seegert D. The IL-6/sIL-6R complex as a novel target for therapeutic approaches. Expert Opin Ther Targets 2007; 11:613 - 624
- Kotake S, Sato K, Kim KJ, Takajashi N, Udagawa N, Nakamura I, et al. Interleukin-6 and soluble interleukin-6 receptors in the synovial fluids from rheumatoid arthritis patients are responsible for osteoclast-like cell formation. J Bone Miner Res 1996; 11:88 - 95
- Lainez B, Fernandez-Reeal JM, Romero X, Esplugues E, Canete JD, Ricart W, et al. Identification and characterization of a novel spliced variant that encodes human soluble tumor necrosis factor receptor 2. Int Immunol 2004; 16:169 - 177
- Graham RR, Kozyrev SV, Baechler EC, Prasad Linga Reddy MV, Plenge RM, Bauer JW, et al. A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nat Genet 2006; 38:550 - 555
- Yasuda S, Stevens RL, Terada T, Takeda M, Hashimoto T, Fukae J, et al. Defective expression of Ras Guanyl Nucleotide-Releasing protein 1 in a subset of patients with systemic lupus erythematosus. J Immunol 2007; 179:4890 - 4900
- Hitomi Y, Tsuchiya N, Kawasaki A, Ohashi J, Suzuki T, Kyogoku X, et al. CD72 polymorphisms associated with alternative splicing modify susceptibility to human systemic lupus erythematosus through epistatic interaction with FCGR2B. Hum Mol Gen 2004; 13:2907 - 2917
- Wei C-C, Chang M-S. A novel transcript of mouse interleukin-20 receptor acts on glomerular mesangial cells as an aggravating factor in lupus nephritis. Genes and Immunity 2008; 9:668 - 679
- Douglas KB, Windels DC, Zhao, Gadeliya AV, Wu H, Kaufmann KM, et al. Complement receptor 2 polymorphisms associated with systemic lupus erythematosus modulate alternative splicing. Genes Immun 2009; 10:457 - 469
- Sakkas LI, Tourtellotte C, Berney S, Myers AR, Platsoucas CD. Increased levels of alternatively spliced interleukin 4 (IL-4δ2) transcripts in peripheral blood mononuclear cells from patients with systemic sclerosis. Clin Diagn Lab Immunol 1999; 6:660 - 664
- Atamas SP, White B. Interleukin 4 in systemic sclerosis: not just an increase. Clin Diagn Lab Immunol 1999; 6:658 - 659
- Hasegawa M, Nakoshi Y, Muraki M, Sudo A, Kinoshita N, Yoshida T, et al. Expression of large tenascin-C splice variants in synovial fluid of patients with rheumatoid arthritis. J Orthop Res 2007; 25:563 - 568
- Kojo S, Tsutsumi A, Goto D, Sumida T. Low expression levels of soluble CD1d gene in patients with rheumatoid arthritis. J Rheumatol 2003; 30:2524 - 2528
- Croft DR, Dall P, Davies D, Jackson DG, McIntyre P, Kramer IM. Complex CD44 splicing combinations in synovial fibroblasts from arthritic joints. Eur J Immunol 1997; 27:1680 - 1684
- Wibulswas A, Croft D, Pitsillides AA, Bacarese-Hamilton I, McIntyre P, Genot E, et al. Influence of epitopes CD44v3 and CD44c6 in the invasive behavior of fibroblast-like synoviocytes derived from rheumatoid arthritis joints. Arthritis Rheum 2002; 46:2059 - 2064
- Naor D, Nedvetzki S. CD44 in rheumatoid arthritis. Arthritis Res Ther 2003; 5:105 - 115
- Shiozawa K, Hino K, Shiozawa S. Alternatively spliced EDA-containing fibronectin in synovial fluid as a predictor of rheumatoid joint destruction. Rheumatology 2001; 40:739 - 742
- Michel J, Langstein J, Hofstadter F, Schwarz H. A soluble form of CD137 (ILA/4-1BB), a member of the TNF receptor family, is released by activated lymphocytes and is detectable in sera of patients with rheumatoid arthritis. Eur J Immunol 1998; 28:290 - 295
- Vambutas A, DeVoti J, Goldofsky E, Gordon M, Lesser M, Bonagura V. Alternate splicing of interleukin-1 receptor type II (IL1R2) in vitro correlates with clinical glucocorticoid responsiveness in patients with AIED. PLoS ONE 2009; 4:5293
- Qu H-Q, Lu Y, Marchand L, Bacot F, Frechette R, Tessier M-C, et al. Genetic control of alternative splicing in the TAP2 gene: Possible implication in the genetics of type 1 diabetes. Diabetes 2007; 56:270 - 275
- Diez J, Park Y, Zeller M, Brown D, Garza D, Ricordi C, et al. Differential splicing of the IA-2 mRNA in pancreas and lymphoid organs as a permissive genetic mechanism for autoimmunity against the IA-2 type 1 diabetes autoantigen. Diabetes 2001; 50:895 - 900