
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The 12th Annual International Conference on the Critical Assessment of Massive Data Analysis (CAMDA) used data from the massive Japanese Toxicogenomics Project (TGP) to predict drug-induced liver injury (DILI) concern provided by the U.S. Food and Drug Administration (FDA). The challenge was to predict DILI concern by means of gene expression data. Analysis of this high-dimensional toxicogenomic data requires statistical methodologies that can detect the transcriptomic associations with toxicity. We propose an analysis technique that involves sparse principal component analysis to efficiently reduce the dimension of the analysis problem. Sparse principal component variables are composed of groups of expressed genes. Associations between DILI concern and sparse principal component variables were tested and further scrutinized with sparse regression methodology to identify concise transcriptomic structures potentially responsible for and predictive of drug toxicity. Working with a subset of the TGP data with FDA DILI concern classification, we identified 5 transcriptomic structures (sparse principal component variables) statistically associated with DILI concern. The most statistically significant structure consists of the genes ZBTB16, FLVCR2, TNS3, and ASB13. Sparse statistical methods offer a new way to handle analysis issues with massive omic data. Sparse PCA can efficiently extract groups of transcriptomic markers that may indicate drug toxicity.
Abbreviations
| CAMDA | = | Critical Assessment of Massive Data Analysis |
| DEG | = | differentially expressed gene |
| DEPC | = | differentially expressed principal component |
| DILI | = | drug-induced liver injury |
| FARMS | = | Factor Analysis for Robust Microarray Summarization |
| PC | = | principal component |
| PCA | = | principal component analysis |
| TGP | = | Japanese Toxicogenomics Project |
Introduction
The current estimated cost in US dollars to develop a drug is $1.8 billion, but most drugs never make it to market, largely due to toxicity levels deemed unsafe for the human liver.Citation1 As it is unethical to test the effects of compounds in humans, toxicity has classically been tested in animal-based experiments. Hence, conclusions may not be generalizable to the human population, especially when the effects of toxicity are revealed after prolonged exposure. The testing inaccuracy could result in passing unsafe or discarding useful drugs, which hinder public health efforts and squander resources. In an attempt to improve testing accuracy, the field of toxicology has embraced animal and now human “omic” data (genomics, transcriptomics, proteomics, metabolomics) to help identify biological material that predict the toxicity of compounds at an early stage. Therefore, resources are saved from developing drugs that will ultimately fail in the public domain. The use of omic data to inform toxicology is commonly referred to as toxicogenomics and is now the focus of several research initiatives. One such initiative is the Japanese Toxicogenomics Project (TGP).Citation2 With human in vitro, rat in vitro, and rat in vivo experimental models, 131 compounds were applied to liver samples and microarray gene expression profiles were obtained using Affymetrix GeneChip® technology.
To facilitate prediction of toxicity in new drugs, the Food and Drug Administration (FDA) developed a classification system of human drug-induced liver injury (DILI) concern (most, less, and no DILI concern) for drugs currently on the market.Citation3 The drugs they classified had been on market for a minimum of 10 y, allowing sufficient public interaction to obtain updated and realistic DILI concern information, not otherwise attainable due to ethical reasons. This new classification of human-based drug toxicity can be linked to toxicogenomic databases, facilitating the search for omic markers that predict toxicity based on this indicator. The 12th Annual International Conference on Critical Assessment of Massive Data Analysis (CAMDA 2013) linked FDA classification labeling to the TGP data and proposed analysis challenges involving prediction of drug toxicity. Discovering novel biomarkers associated with DILI concern within the TGP data may aid in our understanding of mechanisms of toxicity and enhance our ability to assess the toxicity of new compounds. However, the breadth and complexity of omic data makes analysis challenging and to extract key information it is important to enrich statistical methodology with biological context.Citation4
Commonly, simple statistical methods are used to test associations between drug toxicity and each marker, one at a time, developing a list of top candidate genes. Although this approach is easy to implement, it involves conducting thousands of statistical tests and is prone to spurious associations (i.e., the “multiple testing problem”). Perhaps more concerning is that by testing markers independent of one-another, this analysis does not acknowledge the fact that genes may act in concert to influence a phenotype. Complex networks of omic material will likely be the underlying indicator of drug toxicity. Identifying and characterizing these indicators requires more sophisticated statistical methods. We believe that recent advancement in a new class of so-called sparse statistical methods validates the use of principal component analysis (PCA) as a primary analysis tool to detect these complex networks.
PCA is a commonly used multivariate method for both dimension reduction and data visualization. It assembles a new set of variables, called principal components (PCs), from linear combinations of original variables. These PCs are uncorrelated and ordered by maximal variance, giving the analyst an easier data set to work with if they can interpret what the PCs mean. Unfortunately, this is a major challenge with high-dimensional data, as found in toxicogenomics, since the PCs are formed by linear combinations of all original variables (a weighted sum of 1000 genes, for example) and are uninterpretable. To overcome this limitation, sparse principal component analysis methodsCitation5-7 have been developed by combining PCA with sparse regression methodology.Citation8,9 Sparse PCA restricts principal components to be formed by interpretable linear combinations of smaller (sparse) subsets of the original variables (a weighted sum of 10 genes, for example). Tuning parameters control the level of sparseness induced, making sparse PCA procedures very flexible. Sparse principal components reflect concise and interpretable groups of original variables that remain in the linear combinations.
In this paper, we present an analysis strategy built around using sparse PCA to extract groups of related genes from a toxicogenomic database, testing their associations with drug toxicity. We apply this analysis strategy to subsets of the TGP database with the FDA's classification of human DILI concern as the measure of drug toxicity. We hope to convey that this new class of sparse statistical methods may prove beneficial to toxicogenomic analyses, as they specialize at separating the “signal” from “noise” amidst high-dimensional data.
The rest of our paper is organized as follows. In the Materials and Methods section, we describe the data we used, the essentials of sparse methodology, and our analysis strategy. We then present our Results and provide a Discussion on the potential merits and limitations of sparse methodology in toxicogenomic analyses.
Materials and Methods
Description of Data
Data source
Completed in 2007, the TGP database was the result of a 5-y collaborative effort between government and private companies to obtain gene expression data using Affymetrix GeneChip® technology to characterize the response to various drugs across a variety of experimental units.Citation2 The completed TGP database provided by CAMDA 2013 contains thousands of results from experiments, involving 131 drugs applied at 4 dose levels (control, low, middle, and high) to human and rat in vitro hepatocytes, and administered to rat in vivo with either a single dose or repeated doses. Transcriptomic material was extracted from experimental units at several time points after the drugs had been given. Full details regarding the study design and protocol are given by Uehara et al.Citation2 and complete data, along with the portion we obtained including FDA human DILI concern labels, are available through the CAMDA 2013 conference website (http://dokuwiki.bioinf.jku.at/doku.php).
Samples used
We considered only those drugs with FDA human DILI concern classification; 93 of the 119 drugs tested on human samples, and 101 of the 131 drugs tested on rat samples. We primarily focused on the 93 samples from human in vitro experiments that specifically received high dose levels and had gene expression measured at 8 h; a single subset of this rich database. However, to compare results across different experimental conditions, we also repeated our analysis on another 15 subsets of the database. Anticipating that higher doses result in more robust gene expression measurementsCitation10 and due to many drugs not being administered at low doses in the human in vitro samples, we considered only samples that received middle or high dose levels. Likewise, since measurements of gene expression in the human samples were not taken at 2 h for many drugs, we considered only gene expression values measured at later time points.
Transcriptomic markers used
Human gene expression data were obtained with the Affymetrix GeneChip® Human Genome U133 Plus 2.0 Array and rat gene expression data were obtained with Affymetrix GeneChip® RAE 230A 2.0 Array.Citation2 Although raw gene expression data was available, we chose to use preprocessed data that had undergone batch-effect correction and the Factor Analysis for Robust Microarray Summarization (FARMS),Citation11 that was also provided. A total of 18988 probesets were available for replicate-collapsed human samples, and 12088 probesets were available for replicate-collapsed rat samples. Almost all probesets had gene names provided and all gene names were unique; as such we refer to probesets and genes interchangeably unless the gene name was not provided. We used inter-quartile range to filter genes that did not vary much and kept the top 1000 genes to simplify analysis.
Classification variable for human DILI concern
Human DILI concern (“most,” “less,” and “no” DILI concern in humans) was provided to detect if gene expression measurements differed across DILI classification. However, since only 8 drugs are classified as “no DILI concern,” we reclassified the human DILI concern variable to be binary (“most” vs. “less or no” DILI concern) to obtain a more balanced and simple categorical variable. Of the 93 drugs tested in human tissue, 40 are labeled as “most DILI concern” and 53 are “less or no DII concern” and of the 101 drugs tested in rat specimens, 41 are “most DILI concern” and 60 are “less or no DILI concern.”
Sparse PCA and Sparse Regression Methodology
Here we give an intuitive description of sparse PCA and sparse regression and refer the reader to refs.Citation5-9 for mathematical details.
Given a data matrix X = (X1,X2, …, Xp), where p variables (e.g., probsets or other omic markers) are measured for n observations (samples; drugs), classical PCA constructs linear combinations (weighted sums) from the variables in the form
where Zi is the ith principal component (PC) variable and the vi,j's are the loadings (coefficients; weights) for the jth variable in the ith PC. By utilizing the correlation structure among X's, PCA constructs PCs in such a way that they are uncorrelated, ordered by maximal variance, and the total amount of information (variance) in the new data matrix Z = (Z1,Z2, …, Zmin(n,p)) is equal to the total amount of information in the original data matrix X. We may now choose to analyze the Z's instead of X's, as these defining properties potentially simplify analysis. For example, analyzing uncorrelated Z's allows for easier regression analysis as there is no issues with colinearity. Even more appealing, if n < p then the number of Z's to analyze will be less than the number of X's. Likewise, since Z's are ordered by variance, the first few PCs will typically hold a large portion of the information and we can discard the trailing ones without losing much. For these last two reasons, PCA is an excellent so-called dimension-reduction tool, able to compress large amounts of data into a few important components. In some cases, PCA can be an excellent exploratory tool, since groups of highly correlated X's will bear substantial weight in the same PC and we can detect groups of variables through the magnitude of their loadings (vi,j's). However, since PCs are linear combinations of all X's, this makes them nearly impossible to interpret when dealing with thousands of correlated X's, as is common with high-dimensional toxicogenomics data.
Sparse PCA is an extension to classical PCA that forces the loadings for some X's to be exactly 0, creating sparse linear combinations in the form (for example):
where now the vi,j's that do not have much relation to a PC are removed. This results in a more interpretable set of PCs, having small groups of X's (genes, for example) creating new variables to work with. This is objectively achieved through integrating constrained (sparse) regression methodology to the derivation of PCA. For example, finding classical PCA solutions through the singular value decomposition (SVD) X = UDV′, where V holds all the loadings and Z = UD are the PCs, Witten et al.Citation6 applied constraints to elements of V, shrinking some of them to 0. A so-called tuning parameter (λ) controls the number of loadings that are forced to 0, providing a flexible framework from which to obtain sparse PCs. As a trade-off for acquiring interpretable PCs by introducing sparseness, a proportion of the total information (variance) is lost.
Finally, sparse regression works in a similar way, in that the coefficients estimates (β's) of a regression model of the form
are constrained, shrinking a number of them, dictated by tuning parameters, directly to 0. It has been integrated to many extensions of the linear model, such as generalized linear models, including binary logistic regression.
Analysis Strategy
The novel component of our analysis strategy is the use of sparse PCA to automatically select groups of variables, however we suggest a more broad analysis pipeline as visualized in .
Figure 1. A visual of our analysis strategy applied to human in vitro, high dose, 8 h sampling time data. We begin in the top row (1-a), by conducting a Differentially Expressed Genes (DEGs) analysis on the gene expression matrix; columns represent the 93 samples (40 “most” and 53 “less or no” DILI concern), rows represent 1000 expressed genes. This returns a list of top DEGs (1-b); the genes that are most significantly associated with DILI concern. We then move to the middle row (2-a), using sparse PCA on the same gene expression matrix to obtain new sparse principal component (PC) variables (2-b) to work with; columns for this new data matrix again represent the 93 samples, but rows represent the 93 new sparse PC variables (we have reduced the dimension from 1000 to 93). Then, we conduct a Differentially Expressed PCs (DEPCs) analysis on the PC expression matrix to obtain a list of top DEPCs (2-c); the sparse PCs that are most significantly associated with DILI concern. At this point, we examine the genes that contribute to these DEPCs to makes sense of what the structures mean and make note of those genes in these structures that were also identified as differentially expressed in the DEGs analysis. As a final validation step (3-a), we apply sparse regression to the same 93 sparse PC variables to identify a concise list of sparse PCs that are potentially related to DILI concern (3-b).
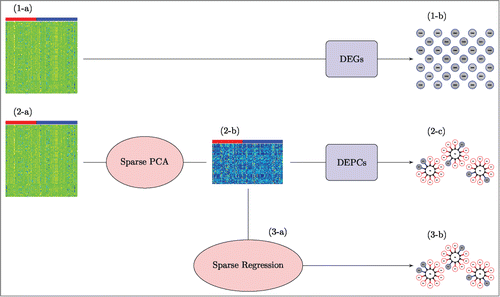
We implemented this analysis for each of the 16 subsets of the TGP data we considered but refer only to the human in vitro, high dose level, 8 h gene expression sampling time subset while explaining the steps in detail. All analysis was conducted with R statistical software (version 3.0.1).Citation12
Step 1: Identify genes independently associated with DILI concern with a Differentially Expressed Gene (DEG) analysis
This step can be visualized with 1-a and 1-b in . Starting with the 1000 most variable genes, we first investigated if any genes are independently associated with DILI concern. Instead of using the two independent samples t test for each gene, we used the moderated t testCitation13 from the R package “limma”Citation14 which yields more conservative P; without this correction, when conducting many tests, suspiciously small standard errors can inflate test statistics. We obtained “moderated t” test statistics with associated P and claimed that genes with P < 0.05 were significantly associated with DILI concern. Additionally, using “limma,” we obtained q values (P adjusted to account for false-discovery rates)Citation15,16 to eliminate genes likely to be claimed falsely significant due to conducting multiple tests. With this list of differentially expressed genes likely to be independently associated with DILI concern, we moved to search for more complex relationships between the gene expression data and DILI concern.
Step 2: Identify groups of genes jointly associated with DILI concern using sparse PCA and a Differentially Expressed Principal Component (DEPC) analysis
This step can be visualized with 2-a, 2-b, and 2-c in . Starting with the 1000 most variable genes, we used the sparse PCA method by Witten et al.Citation6 executed with the R package “PMA”Citation17, to obtain sparse principal component variables. We chose this sparse PCA method over certain other formulationsCitation5,7 as a result from our previous workCitation18 that found it to perform better in computer simulations with high-dimensional data. For each of a range of tuning parameters (λ = 2, 3, 5, 7, 10, 15), we ran sparse PCA and examined the trade-off between adjusted percentage explained varianceCitation18 and sparseness among PCs with the goal to select a tuning parameter that delivered principal components with extremely sparse loading vectors while keeping a large proportion of variance. We chose tuning parameter λ = 3 because while compared with λ = 5, 7, 10, and 15 it resulted in loading vectors that were drastically more sparse at a small cost of information loss and loading vectors were almost as sparse as those from λ = 2 while comparatively keeping a substantial amount of information. Additionally, and important from a practical view, this delivered small and interpretable linear combinations so we could look at the principal components in clear detail. We then statistically tested associations between each sparse PC variable and DILI concern using a permutation approach claiming sparse PCs with P < 0.05 to be significantly associated with DILI concern. With this list of “differentially expressed” sparse PCs (DEPCs) likely to be independently associated with DILI concern, we then recorded and examined the genes that made up the PCs along with their respective loadings in the PC linear combination.
Step 3: Validate DEPCs with Sparse Regression
This step can be visualized with 3-a and 3-b in . We simultaneously entered the sparse principal component variables into a binary logistic regression model framework with DILI concern as the outcome, where the coefficient estimates were obtained with sparse regression methodology; penalized maximum likelihood with a least absolute shrinkage and selection operator (LASSO) penaltyCitation8 with tuning parameter selected via a cross validation procedure that maintains good prediction from the model, executed with the R package “glmnet.”Citation19 From this sparse regression model, we obtained the list of sparse PC variables that remained, expecting overlap with the list of sparse PCs identified to be significantly related to DILI concern in Step 2, then recorded and examined the genes and loadings in these PCs. Sparse PCs found both in Step 2 and Step 3 were deemed to be most likely to be associated with DILI concern.
Results
We now present the results from our analysis strategy, reporting details on the human in vitro, high dose, 8 h gene expression sampling time subset but also including summaries of findings for the other 15 subsets.
Of the 1000 genes, 54 had significantly different expression between “most” and “less or no” DILI concern samples, according to the moderated t tests with P < 0.05. displays their gene names, effect size, and associated P values.
Table 1. Differentially expressed genes (DEGs; independently associated with DILI concern) from our analysis of the human in vitro, high dose, 8 h gene expression sampling time subset
None of these 54 genes remained statistically significant when using the FDR-adjusted P value (q value) with cut-off q < 0.10. displays counts of the number of DEGs found in each of the 16 subsets we analyzed.
Table 2. Counts of DEGs across all 16 subsets analyzed
The rat in vivo samples receiving single dose at middle dose levels with gene expression measured at 9 h had a notably large amount of DEGs compared with the rest; 228 with 202 remaining statistically significant after the more strict FDR-adjustment.
Applying sparse PCA with tuning parameter λ = 3 to the gene expression data, we obtained the 93 [min(n = 93, p = 1000)] sparse principal components. The minimum, median, and maximum number of genes that contributed to a sparse principal component with non-zero loadings was 11, 18, and 33, respectively. Compared with classical PCA, which would return principal components built from all 1000 genes, this is a substantial amount of sparseness and, therefore, interpretability gained. As a sacrifice for simplifying the data in this way, we lost 29.97% of total variance (information) contained in the original 1000 genes. Considering that sparse PCA simplified our focus to a median of just 18 (1.8% of the 1000) genes per component, therefore giving us groups of genes to explore, and reduced our analysis burden from 1000 gene tests to 93 principal component tests, we consider this a massive gain for a comparably small loss. Similar data reduction statistics were observed for the remaining 15 subsets.
Of the 93 sparse principal component variables, 5 had significantly different cumulative expression values between “most” and “less or no” DILI concern samples, according to the permutation test with P < 0.05. provides a summary of these DEPCs, including P, a list of contributing genes, and how many of the contributing genes were found in the DEG analysis.
Table 3. Differentially expressed PCs (DEPCs; associated with DILI concern) from our analysis of the human in vitro, high dose, 8 h gene expression sampling time subset
provides a visual representation of the top 3 of these PCs, showing the different types of groups of genes that are associated with DILI concern, in terms of size and composition.
Figure 2. A visual display for the top 3 differentially expressed (most associated with DILI concern) sparse principal components (DEPCs) from our analysis of the human in vitro, high dose, 8 h gene expression sampling time subset. Larger central circles represent the principal components. Attached to each are the genes that form the linear combinations; probesets (gene names) and loading values are inside the outer circles. Shaded circles represent genes that were found to be independently associated with DILI concern (DEGs), whereas non-shaded circles contain genes that were not. PC15 might bring forth a network of trancriptomic material that is associated with DILI concern, not otherwise being found with more simple statistical tests. PC13 shows us that some marginally associated genes behave similarly.
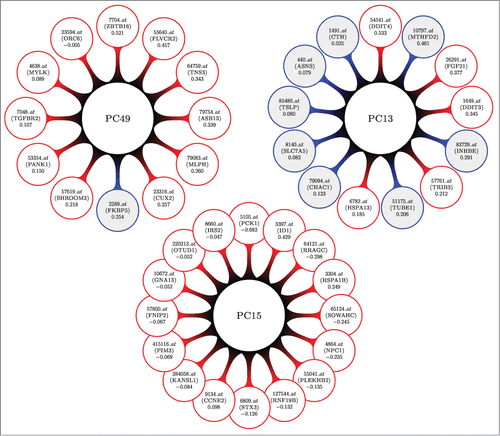
PC49 consists predominantly of probesets (genes), 7704_at (ZBTB16), 55640_at (FLVCR2), 64759_at (TNS3), and 79754_at (ASB13), with lesser contribution from eight more correlated genes, meaning these may have a joint relationship with drug toxicity. One of these eight, 2289_at (FKBP5), was identified as significantly associated with DILI concern in the DEG analysis. PC15 is a slightly larger network comprised entirely of genes which were not identified in the DEG analysis, meaning these genes might only be identified when accumulated into a composite variable like this sparse principal component. PC15 is driven by probesets (genes) 5105_at (PCK1), 3397_at (ID1), and 64121_at (RRAGC). In contrast, 8 out of 13 genes influencing PC13 had independent associations with DILI concern. Its top 4 weighted probesets (genes), 54541_at (DDIT4), 10797_at (MTHFD2), 26291_at (FGF21), and 1649_at (DDIT3), had only one DEG, meaning these genes are likely related to drug toxicity although they did not independently surface. reports the number of sparse PCs that were significantly related to DILI concern across all 16 subsets of data we analyzed.
Table 4. Counts of DEPCs across all 16 subsets analyzed. The total number of DEPCs, along with the number of those which are upregulated (“most DILI concern” has larger PC cumulative expression values than “Less or No DILI concern”) in brackets and those which are downregulated in square brackets
Lastly, sparse regression was used to narrow down a more concise list of DEPCs. Entering the 93 sparse PCs, the model selected PC49 and PC15. These are the top 2 sparse PCs we identified to be most significantly associated with human DILI concern in the DEPCs analysis, bringing some statistical validation to these findings.
Discussion
In this paper, we identified sparse PCs that are differentially expressed between groups of “most” and “less or no” DILI concern, providing small networks of genes for further study in relation to drug toxicity. Apart from being able to investigate groups of variables, there are several components that make sparse PCA attractive. Since the number of principal components returned is min(n,p) (i.e., minimum of n samples and p variables), it will always reduce the number of variables to analyze when applied to high-dimensional (n < p) data. This is greatly beneficial for n < < p data, as we observed with the TGP data set; reducing from 1000 to 93 tests. By reducing the dimensional complexity of the analysis this strategy also reduces the burden of multiple testing, but it does not eradicate the issue and necessary adjustments should be made before concluding if PCs are statistically significant. In contrast to classical PCs, we argue that sparse PCs are interpretable since they are linear combinations consisting of small subsets of genes; however biological context is still required to interpret their values. Built from gene expression data, the sparse PCs in this paper are weighted sums of gene expression values, meaning they can be interpreted as accumulated gene expression across the genes involved. Within the same PC, genes with loading values that have opposite signs are negatively correlated, giving additional insight into structures between genes in the same PC. Since our method returns composite variables, as opposed to simply reporting groups of similar genes as with cluster analysis,Citation20 this allows for statistical testing of joint associations. Since PCA methods are unsupervised (meaning they are not informed by the outcome of interest, such as DILI concern), it is reasonable to expect to find PCs built exclusively from genes that are not differentially expressed. Any of these types of group structures that are found to be associated with the variable of interest potentially offer a brand new source of transcriptomic material for researchers to explore. PC15 in our analysis of the TGP data is an example of this.
We have some cautionary notes regarding the selection of tuning parameters when using sparse PCA to simplify data in search of groups of genes, as it can have major effects on characteristics of the sparse principal components obtained. By choosing λ = 3, for example, we committed to identifying groups of genes of a specific size; a median of 18 genes contributed to building the sparse principal components we obtained. For the purpose of demonstrating immense sparseness, this tuning parameter was a good choice. However, perhaps the true transcriptomic structure underlying the TGP gene expression data involves hundreds of correlated genes expressing similarly and as such, perhaps choosing a less sparse tuning parameter would more accurately capture this truth. This subjective choice of tuning parameter is analogous to restricting the number of clusters, k, in the k-means clustering algorithm. Prediction-based cross-validation methods are available to guide the choice of tuning parametersCitation6 and are typically the default methods built into packages.Citation17 BonnerCitation18 suggested to visually inspect the variance-covariance structure of data to observe blocking of variables (genes), but this strategy has yet to be tested with rigour or under realistic data conditions.
The analysis strategy we proposed is a practical way to explore multivariate patterns in large genomic data and suggest if they are worth pursuing. Sparse methodology has many extensions to a variety of data analysis situations and it has potential to benefit research in toxicogenomics. The R scripts and analysis pipeline we used in this paper are available upon request.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
JB would like to acknowledge funding from the Natural Sciences and Engineering Research Council of Canada (NSERC) and Canadian Institutes of Health Research (CIHR) (grant number 84392). JB is the inaugural holder of the John D. Cameron Endowed Chair in the Genetic Determinants of Chronic Diseases, Department of Clinical Epidemiology and Biostatistics, McMaster University.
References
- Taboureau O, Hersey A, Audouze K, Gautier L, Jacobsen U, Akhtar R, Atkinson F, Overington J, Brunak S. Toxicogenomics Investigation Under the eTOX Project. Pharmacogenomics & Pharmacoproteomics 2012; S7.
- Uehara T, Ono A, Maruyama T, Kato I, Yamada H, Ohno Y, Urushidani T. The Japanese toxicogenomics project: application of toxicogenomics. Mol Nutr Food Res 2010; 54:218-27; PMID:20041446; http://dx.doi.org/10.1002/mnfr.200900169
- Chen M, Vijay V, Shi Q, Liu Z, Fang H, Tong W. FDA-approved drug labeling for the study of drug-induced liver injury. Drug Discov Today 2011; 16:697-703; PMID:21624500; http://dx.doi.org/10.1016/j.drudis.2011.05.007
- Afshari CA, Hamadeh HK, Bushel PR. The evolution of bioinformatics in toxicology: advancing toxicogenomics. Toxicol Sci 2011; 120(Suppl 1):S225-37; PMID:21177775; http://dx.doi.org/10.1093/toxsci/kfq373
- Zou H, Hastie T, Tibshirani R. Sparse Principal Component Analysis. J Comput Graph Stat 2006; 15:265-86; http://dx.doi.org/10.1198/106186006X113430
- Witten DM, Tibshirani R, Hastie T. A penalized matrix decomposition, with applications to sparse principal components and canonical correlation analysis. Biostatistics 2009; 10:515-34; PMID:19377034; http://dx.doi.org/10.1093/biostatistics/kxp008
- Lee D, Lee W, Lee Y, Pawitan Y. Super-sparse principal component analyses for high-throughput genomic data. BMC Bioinformatics 2010; 11:296-305; PMID:20525176; http://dx.doi.org/10.1186/1471-2105-11-296
- Tibshirani R. Regression shrinkage and Selection via the Lasso. J R Stat Soc, B 1996; 58:267-88.
- Zou H, Hastie T. Regularization and variable selection via the elastic net. J R Stat Soc, B 2005; 67:301-20; http://dx.doi.org/10.1111/j.1467-9868.2005.00503.x
- McMillian M, Nie A, Parker JB, Leone A, Kemmerer M, Bryant S, Herlich J, Yieh L, Bittner A, Liu X, et al. Drug-induced oxidative stress in rat liver from a toxicogenomics perspective. Toxicol Appl Pharmacol 2005; 207(Suppl):171-8; PMID:15982685; http://dx.doi.org/10.1016/j.taap.2005.02.031
- Hochreiter S, Clevert D-A, Obermayer K. A new summarization method for Affymetrix probe level data. Bioinformatics 2006; 22:943-9; PMID:16473874; http://dx.doi.org/10.1093/bioinformatics/btl033
- R Core Team. (2013). R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. URL http://www.R-project.org/
- Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 2004; 3:e3; PMID:16646809; http://dx.doi.org/10.2202/1544-6115.1027
- Smyth GK. Limma: linear models for microarray data. In: Bioinformatics and Computational Biology Solutions using R and Bioconductor 2005, R. Gentleman, V. Carey, S. Dudoit, R. Irizarry, W. Huber (eds.), Springer, New York, pages 397-420.
- Benjamin Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc, B 1995; 57:289-300.
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A 2003; 100:9440-5; PMID:12883005; http://dx.doi.org/10.1073/pnas.1530509100
- Witten D. Tibshirani Rob, Gross S, Narasimhan B. PMA: Penalized Multivariate Analysis. R package version 1.0.9 (2013). http://CRAN.R-project.org/package=PMA
- Bonner A. Sparse principal component analysis for high-dimensional data: a comparative study. Open Access Dissertations and Theses - McMaster 2012
- Friedman J, Hastie T, Tibshirani R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J Stat Softw 2010; 33:1-22; PMID:20808728
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A 1998; 95:14863-8; PMID:9843981; http://dx.doi.org/10.1073/pnas.95.25.14863