Abstract
Blood-brain barrier (BBB) regulates transport of various molecules and maintains brain homeostasis. Perturbed intracellular Ca2+ homeostasis and BBB damage have been implicated in the pathogenesis of Alzheimer disease (AD). Although receptor for advanced glycation end products (RAGE) is known to mediate Aβ transcytosis across the BBB, molecular mechanisms underlying Aβ-RAGE interaction-induced BBB alterations are largely unknown. We found enhanced permeability, decreased zonula occludin-1 (ZO-1) expression and increased intracellular calcium and MMP secretion in endothelial cells exposed to Aβ1–42. Aβ-induced changes in ZO-1 were attenuated by neutralizing antibodies against RAGE and inhibitors of calcineurin (CaN) and MMPs, suggesting that Aβ-RAGE interactions disrupt tight junction proteins via the Ca2+-CaN pathway. We also found disrupted microvessels near Aβ plaque-deposited areas, elevated RAGE expression and enhanced MMP secretion in microvessels of the brains of 5XFAD mice, an animal model of AD. These results identify a potential molecular pathway underlying Aβ-RAGE interaction-induced breakage of BBB integrity.
Alzheimer disease (AD) is a progressive neurodegenerative disorder characterized by the accumulation of amyloid β-peptide (Aβ) in the central nervous system (CNS), the presence of hyperphosphorylated tau filaments and cerebrovascular changes that lead to cerebral amyloid angiopathy (CAA).Citation1,Citation2 The accumulation of Aβ peptides is believed to be an early and causative event in cerebrovascular alterations.Citation3 Previous reports have shown that alteration of microvascular permeability and disruption of the BBB are detected in the brains of AD subjects and are the major event of AD.Citation4,Citation5 Yet, the deposition of Aβ aggregates in cerebral blood vessels and the brain is poorly understood, and the mechanisms that underlie the response to changes in permeability are not clear.
The blood brain barrier (BBB) is a specialized barrier that controls the transport of various molecules and maintains the integrity of brain by restricting permeability across brain endothelium.Citation6 Tight junctions (TJs) between endothelial cells in brain capillaries are the most important structural elements of the BBB. As a component of the TJ, zonula occluden protein-1 (ZO-1) was initially identified in the BBB and associated with TJ integrity.Citation7 ZO-1 is a peripheral membrane protein that localizes along blood vessels to form the BBB in the brain parenchyma. ZO-1 binds directly to a wide variety of cellular proteins, such as occludin and claudins in vitro,Citation8 and orchestrates the formation of TJ complexes. Because ZO-1 is sufficient to mitigate alterations in TJ integrity, we examined Aβ-induced structural changes in ZO-1.
BBB regulates the entry of plasma-derived Aβ into the CNS and clears brain-derived Aβ through the receptor for advanced glycation end products (RAGE) and low-density lipoprotein receptor-related protein (LRP), respectively.Citation9-Citation11 In previous reports, AD patients or AD mouse models showed increased levels of free Aβ in plasma.Citation12,Citation13 Through these important clues, we suggest that Aβ may disrupt the TJ of BBB via interaction with RAGE as a specific mediator. It has been known that calcium influx is induced by Aβ in the cells,Citation14,Citation15 and the elevated intracellular calcium leads to alteration of TJs as well as induction of matrix metalloproteinases (MMPs) expression.Citation16,Citation17 In this study, using neutralizing anti-RAGE and specific inhibitors of calcineurin (CaN) and MMPs, we found that Aβ−induced TJ disruptions are mediated by RAGE via intracellular Ca2+-CaN signaling and MMP secretion. In addition, we found that alterations of cerebral capillaries, RAGE expression and TJ structural changes have a causal relationship in 5XFAD mouse brains, AD animal model.
To assess the mechanisms by which Aβ1−42 disrupts TJs and induces structural alterations in ZO-1 in monolayer culture of bEnd.3 cells, BBB permeability was examined with various methods. We confirmed that 5 μM Aβ1−42 induced structural alteration and reduced the protein level of other TJ proteins such as claudin-5 and occludin as well as ZO-1. Also, 5 μM Aβ1−42 increased the amount of diffused FITC-dextran (FD-40) in transwell system of bEnd.3 cells, suggesting that Aβ1−42 could open paracellular pathway due to disrupt TJ integrity. To examine the mechanism of Aβ1−42 -induced disruption of TJ integrity, RAGE was monitored because RAGE is well known receptor for Aβ in the BBB. Neutralizing antibody against the extracellular domain of RAGE effectively blocked Aβ1–42-induced perturbations in ZO-1 distribution, supporting that Aβ-RAGE interactions are critical for TJ integrity. We confirmed that increased intracellular calcium levels with RAGE and activated MMPs through the CaN pathway are associated with Aβ1–42 induced alterations in TJs in bEnd.3 cells as demonstrated by treatment with CaN and MMP inhibitors. Moreover, these events were mitigated by a neutralizing antibody against RAGE, suggesting that disruption of the BBB by Aβ1–42 is initiated by an interaction between Aβ1–42 and RAGE, followed by intracellular signaling cascades in bEnd.3 cells. We have observed that RAGE affects Aβ-induced calcium influxCitation18 and that the influx is sustained during Aβ treatment in present study. Thus, we suggest that RAGE not only transports Aβ into the brain but also mediates Aβ-induced signaling, suggesting a new function of TJ disruption in endothelial cells. Although previous reports suggest that Aβ induced cell death on human and rat cerebral endothelial cells,Citation19,Citation20 cell death was unaffected by Aβ in this culture system. Since unaltered morphology of cells in Aβ-treated group, which is comparable to vehicle (DMSO)-treated group was observed, we think that integrity of cytoskeletal proteins such as ZO-1, occludin and claudin-5 are altered under this situation without affecting cell death.
GM6001 is known to prevent the conversion of pro-MMPs to active forms of matrix-degrading MMPs.Citation21 FK506, which is widely used to prevent a CaN upregulation by Aβ, reduces the accumulation of Aβ and mitigates gliosis and CaN activity in the brain of APP/PS1 mouse model of AD.Citation22 Little is known about the effect of GM6001 and FK506 on Aβ-induced alteration of TJs and BBB permeability. Importantly, both GM6001 and FK506 declined Aβ-induced ZO-1 alteration and BBB permeability, suggesting that both CaN and MMPs are able to influence BBB maintenance. When we analyzed an altered ZO-1 distribution by immunostaining with ZO-1 specific antibody, both GM6001 and FK506 increased the stability of TJs in Aβ-treated cells. Earlier studies have established the role of calcium in maintaining the normal TJ morphology.Citation17,Citation23 Aβ induces intracellular Ca2+-CaN signaling in the cellsCitation24,Citation25 and disrupted calcium homeostasis has been reported in the brains of AD patients.Citation26 Therefore, it is possible that effects of FK506 and altered ZO-1 distribution by Aβ have a causal relationship. Several lines of evidence, including our previous report, have shown that RAGE levels rise with age in rodents and humans.Citation18,Citation27 Furthermore, RAGE expression in neurons and microvascular endothelial cells in human brain is increased on treatment with Aβ.Citation18,Citation28 The present study demonstrated that Aβ1–42 decreased ZO-1 levels in full-length human RAGE-overexpressing bEnd.3 cells to a greater extent than control cells, suggesting that RAGE mediates Aβ1–42–induced changes in ZO-1 localization and protein level.
To confirm these data in animal model of AD, we used 5XFAD mice which show massive Aβ accumulation in the brain with neuronal loss at two months old and behavioral abnormality at six to eight months old.Citation29 By confocal microscopy and the super-resolution Structured Illumination Microscopy (SIM, Nikon), we observed that cerebral capillaries of 5XFAD mice were more disconnected and damaged near the deposited area of amyloid plaques while littermates showed a whole, enclosed thick and strong vessel. At the same time, RAGE expression in cerebral capillaries were upregulated in 5XFAD mouse brains, suggesting that the increased Aβ-RAGE interactions could amplify TJ alterations in BBB as confirmed in bEnd.3 cells. Disrupted BBB passes many toxic molecules including Aβ itself, following acceleration of neuronal cell death in the brain. In addition, EM study showed alterations in TJ morphology from 5XFAD mouse brains. This result supports a possibility of BBB breakdown in AD brains. Although we did not perform the experiments with FK506 or GM6001-injected 5XFAD mice, we performed only one dose (5 mg/kg) IP injection with FK506 in three mice for two months (every two day injection) as a preliminary experiment. We could find the tendency of increase of GLUT-1 protein level by FK506 treated mice compared with vehicle injected mice. We need to perform these experiments with various doses and more mice samples with a long-term experimental plan as a following paper. Previous reports demonstrated amyloid plaque burden is reduced in FK506-injected APPswe/PS1dE9 mouse brain and GM6001 reduces oxidative stress associated CAA in the same animal model.Citation22,Citation30 Taken together, these results indicate that Aβ1–42 treatment induces RAGE expression and that the interaction between Aβ and RAGE triggers an intracellular signaling cascade that disrupts TJs, leading to breakdown of BBB integrity. We confirmed that Aβ-induced TJ breakdown occurred by monitoring increases in intracellular calcium, CaN and MMPs using potent inhibitors. Furthermore, BBB disruption was confirmed in vivo AD animal model. On the basis of these results, we conclude that the Aβ-RAGE-CaN-MMP cascade () is an important mechanism of BBB disruption and AD pathogenesis and an excellent target for treating AD.
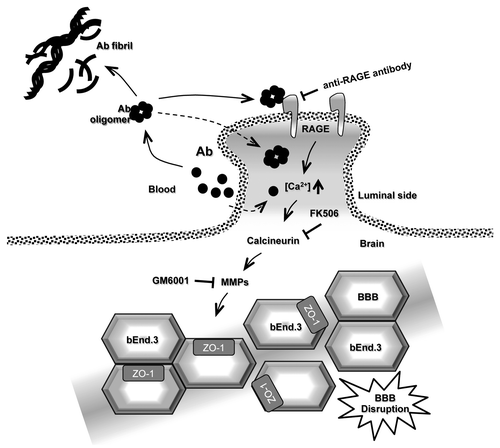
Figure 1. Proposed model of Aβ1–42 -induced TJ disruption in the BBB. Aβ peptide is a proteolytic product generated by the sequential cleavage of amyloid precursor protein (APP). Aβ tends to aggregate and produce Aβ oligomers and fibrils. Aβ in blood interacts with RAGE on endothelial cells of the BBB. This interaction induces intracellular calcium influx into the cell and triggers calcium signaling and CaN activation. The cells increase MMP secretion, resulting in TJ breakdown and increase for BBB permeability. Dotted line indicates luminal side between blood and brain.
| Abbreviations: | ||
| Aβ | = | amyloid-β peptide |
| AD | = | Alzheimer disease |
| BBB | = | blood brain barrier |
| CAA | = | cerebral amyloid angiopathy |
| CaN | = | calcineurin |
| CNS | = | central nervous system |
| EM | = | electron microscopy |
| FD-40 | = | FITC-dextran 40,000 kDa |
| GLUT1 | = | glucose transporter type-1 |
| LRP | = | low-density lipoprotein receptor-related protein |
| MMP | = | matrix metalloproteinase |
| RAGE | = | receptor for advanced glycation end products |
| SIM | = | structured illumination microscopy |
| TJ | = | tight junction |
| ZO-1 | = | zonula occludens-1 |
Acknowledgments
This work was supported by grants from National Research Foundation (2012R1A2A1A01002881, 2008–05943), Medical Research Center (2011–0030738), World class University (R32–10084) and Korean National Institute of Health Road R and D program project (A092058)
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Nagy Zs, Esiri MM, Joachim C, Jobst KA, Morris JH, King EM, et al. Comparison of pathological diagnostic criteria for Alzheimer disease. Alzheimer Dis Assoc Disord 1998; 12:182 - 9; http://dx.doi.org/10.1097/00002093-199809000-00010; PMID: 9772021
- Ellis RJ, Olichney JM, Thal LJ, Mirra SS, Morris JC, Beekly D, et al. Cerebral amyloid angiopathy in the brains of patients with Alzheimer’s disease: the CERAD experience, Part XV. Neurology 1996; 46:1592 - 6; http://dx.doi.org/10.1212/WNL.46.6.1592; PMID: 8649554
- Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev 2001; 81:741 - 66; PMID: 11274343
- Claudio L. Ultrastructural features of the blood-brain barrier in biopsy tissue from Alzheimer’s disease patients. Acta Neuropathol 1996; 91:6 - 14; http://dx.doi.org/10.1007/s004010050386; PMID: 8773140
- Heyman A, Fillenbaum GG, Welsh-Bohmer KA, Gearing M, Mirra SS, Mohs RC, et al. Cerebral infarcts in patients with autopsy-proven Alzheimer’s disease: CERAD, part XVIII. Consortium to Establish a Registry for Alzheimer’s Disease. Neurology 1998; 51:159 - 62; http://dx.doi.org/10.1212/WNL.51.1.159; PMID: 9674796
- Abbott NJ, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci 2006; 7:41 - 53; http://dx.doi.org/10.1038/nrn1824; PMID: 16371949
- Watson PM, Anderson JM, Vanltallie CM, Doctrow SR. The tight-junction-specific protein ZO-1 is a component of the human and rat blood-brain barriers. Neurosci Lett 1991; 129:6 - 10; http://dx.doi.org/10.1016/0304-3940(91)90708-2; PMID: 1922971
- Fanning AS, Jameson BJ, Jesaitis LA, Anderson JM. The tight junction protein ZO-1 establishes a link between the transmembrane protein occludin and the actin cytoskeleton. J Biol Chem 1998; 273:29745 - 53; http://dx.doi.org/10.1074/jbc.273.45.29745; PMID: 9792688
- Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 1996; 382:685 - 91; http://dx.doi.org/10.1038/382685a0; PMID: 8751438
- Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med 2003; 9:907 - 13; http://dx.doi.org/10.1038/nm890; PMID: 12808450
- Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, et al. Clearance of Alzheimer’s amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest 2000; 106:1489 - 99; http://dx.doi.org/10.1172/JCI10498; PMID: 11120756
- Matsubara E, Ghiso J, Frangione B, Amari M, Tomidokoro Y, Ikeda Y, et al. Lipoprotein-free amyloidogenic peptides in plasma are elevated in patients with sporadic Alzheimer’s disease and Down’s syndrome. Ann Neurol 1999; 45:537 - 41; http://dx.doi.org/10.1002/1531-8249(199904)45:4<537::AID-ANA20>3.0.CO;2-2; PMID: 10211483
- Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J Neurosci 2001; 21:372 - 81; PMID: 11160418
- Kawahara M, Kuroda Y, Arispe N, Rojas E. Alzheimer’s beta-amyloid, human islet amylin, and prion protein fragment evoke intracellular free calcium elevations by a common mechanism in a hypothalamic GnRH neuronal cell line. J Biol Chem 2000; 275:14077 - 83; http://dx.doi.org/10.1074/jbc.275.19.14077; PMID: 10799482
- Kagan BL, Hirakura Y, Azimov R, Azimova R, Lin MC. The channel hypothesis of Alzheimer’s disease: current status. Peptides 2002; 23:1311 - 5; http://dx.doi.org/10.1016/S0196-9781(02)00067-0; PMID: 12128087
- Bond M, Fabunmi RP, Baker AH, Newby AC. Synergistic upregulation of metalloproteinase-9 by growth factors and inflammatory cytokines: an absolute requirement for transcription factor NF-kappa B. FEBS Lett 1998; 435:29 - 34; http://dx.doi.org/10.1016/S0014-5793(98)01034-5; PMID: 9755853
- Stuart RO, Sun A, Bush KT, Nigam SK. Dependence of epithelial intercellular junction biogenesis on thapsigargin-sensitive intracellular calcium stores. J Biol Chem 1996; 271:13636 - 41; http://dx.doi.org/10.1074/jbc.271.23.13636; PMID: 8662885
- Cho HJ, Son SM, Jin SM, Hong HS, Shin DH, Kim SJ, et al. RAGE regulates BACE1 and Abeta generation via NFAT1 activation in Alzheimer’s disease animal model. FASEB J 2009; 23:2639 - 49; http://dx.doi.org/10.1096/fj.08-126383; PMID: 19332646
- Miravalle L, Tokuda T, Chiarle R, Giaccone G, Bugiani O, Tagliavini F, et al. Substitutions at codon 22 of Alzheimer’s abeta peptide induce diverse conformational changes and apoptotic effects in human cerebral endothelial cells. J Biol Chem 2000; 275:27110 - 6; PMID: 10821838
- Folin M, Baiguera S, Tommasini M, Guidolin D, Conconi MT, De Carlo E, et al. Effects of beta-amyloid on rat neuromicrovascular endothelial cells cultured in vitro. Int J Mol Med 2005; 15:929 - 35; PMID: 15870895
- Restituito S, Khatri L, Ninan I, Mathews PM, Liu X, Weinberg RJ, et al. Synaptic autoregulation by metalloproteases and γ-secretase. J Neurosci 2011; 31:12083 - 93; http://dx.doi.org/10.1523/JNEUROSCI.2513-11.2011; PMID: 21865451
- Hong HS, Hwang JY, Son SM, Kim YH, Moon M, Inhee MJ. FK506 reduces amyloid plaque burden and induces MMP-9 in AβPP/PS1 double transgenic mice. J Alzheimers Dis 2010; 22:97 - 105; PMID: 20847451
- Gonzalez-Mariscal L, Contreras RG, Bolívar JJ, Ponce A, Chávez De Ramirez B, Cereijido M. Role of calcium in tight junction formation between epithelial cells. Am J Physiol 1990; 259:C978 - 86; PMID: 2124417
- Liu F, Grundke-Iqbal I, Iqbal K, Oda Y, Tomizawa K, Gong CX. Truncation and activation of calcineurin A by calpain I in Alzheimer disease brain. J Biol Chem 2005; 280:37755 - 62; http://dx.doi.org/10.1074/jbc.M507475200; PMID: 16150694
- Agostinho P, Lopes JP, Velez Z, Oliveira CR. Overactivation of calcineurin induced by amyloid-beta and prion proteins. Neurochem Int 2008; 52:1226 - 33; http://dx.doi.org/10.1016/j.neuint.2008.01.005; PMID: 18295934
- Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE. beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci 1992; 12:376 - 89; PMID: 1346802
- Simm A, Casselmann C, Schubert A, Hofmann S, Reimann A, Silber RE. Age associated changes of AGE-receptor expression: RAGE upregulation is associated with human heart dysfunction. Exp Gerontol 2004; 39:407 - 13; http://dx.doi.org/10.1016/j.exger.2003.12.006; PMID: 15036400
- Li M, Shang DS, Zhao WD, Tian L, Li B, Fang WG, et al. Amyloid beta interaction with receptor for advanced glycation end products up-regulates brain endothelial CCR5 expression and promotes T cells crossing the blood-brain barrier. J Immunol 2009; 182:5778 - 88; http://dx.doi.org/10.4049/jimmunol.0803013; PMID: 19380826
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci 2006; 26:10129 - 40; http://dx.doi.org/10.1523/JNEUROSCI.1202-06.2006; PMID: 17021169
- Garcia-Alloza M, Prada C, Lattarulo C, Fine S, Borrelli LA, Betensky R, et al. Matrix metalloproteinase inhibition reduces oxidative stress associated with cerebral amyloid angiopathy in vivo in transgenic mice. J Neurochem 2009; 109:1636 - 47; http://dx.doi.org/10.1111/j.1471-4159.2009.06096.x; PMID: 19457117