Abstract
The hormone HGF regulates morphogenesis and regeneration of multiple organs and increased HGF signaling is strongly associated with metastatic cancer. At the cellular level, one of the distinct effects of HGF is the de-stabilization of cell-cell junctions. Several molecular mechanisms have been shown to be involved that mostly culminate at the E-cadherin adhesion complex. One of the key determinants in HGF-driven morphological changes is the actomyosin cytoskeleton whose organization and physical parameters changes upon stimulation. Here we have investigated how HGF affects the different actomyosin-associated cell-cell junction complexes, Nectin Junctions, Adherens Junctions and Tight Junctions in MDCK cells. We find that components of all complexes stay present at cell-cell contacts until their physical dissociation. We find that at cell-cell junctions, the mobility of Claudin-3, but not that of other cell-cell adhesion receptors, is affected by HGF. This depends on tyrosine residues that likely affect PDZ-domain interactions at the C-terminal tail of Claudin-3, although their phosphorylation is not directly regulated by HGF. Thus we uncovered Claudins as novel targets of HGF signaling at cell-cell junctions.
Introduction
Hepatocyte Growth Factor (HGF) signaling is involved in processes such as cell motility, wound healing, angiogenesis, growth and morphogenesis.Citation1,Citation2 HGF signaling is increased in many different types of cancer, either by increased HGF secretion from stromal fibroblasts or by upregulation of its receptor tyrosine kinase c-MET in tumor cells. c-MET/HGF high tumors often show an invasive phenotype and c-MET/HGF expression in tumors strongly correlates with the development of metastasis.Citation3,Citation4 Because of this, c-MET signaling is a target for anti-cancer drug development by a number of pharmaceutical companies (reviewed inCitation5). The exact mechanisms by which HGF exerts its distinct metastatic effects are still under investigation and may generate additional targets for such drug development. Besides its role in cancer, HGF is critically involved in the morphogenesis and regeneration after damage of multiple organsCitation3 and, therefore, its downstream signaling processes and mechanisms are also very relevant for the current advances in tissue regeneration for clinical purposes.
In 2D epithelial cell culture, HGF-induced signaling through the c-MET receptor results in disruption of cell-cell junctions and subsequent cell scattering. Because loss of epithelial cell-cell adhesion is a key process in tumor metastasis, this in vitro model system has been widely used for the identification of signaling intermediates involved. Classical growth factor induced signaling pathways like Ras-ERK2 and PI3K signaling are clearly involved,Citation6 but what explains the distinct effects of HGF that are not common to other growth factors, on cell-cell junction regulation is not clear. One cellular system that appears to be of particular importance is the actomyosin cytoskeleton. For instance, it was shown that the efficiency of cell-cell disruption in MDCK cells depends on an integrin-dependent increase in tension of the actomyosin cytoskeleton.Citation7 Furthermore, a loss of apical actomyosin structure due to the uncoupling of Myosin VI from E-cadherin was observed after HGF stimulation in colon carcinoma cells, which resulted in a fragmented Zonula Adherens (ZA) and altered cell-cell adhesion.Citation8 Thus, the connections between actomyosin and cell-cell junction complexes may serve as sites of junction regulation that may be specifically affected by HGF signaling.
The best studied actomyosin-connected cell-cell adhesion complex in this context is the E-cadherin complex, which is the central adhesion receptor in Adherens Junctions (AJs). It is clear that HGF induces alterations in this complex through increases in actomyosin contraction and organization.Citation7,Citation9 Several other actomyosin associated adhesion complexes are prominent in epithelial cells, however, whose regulation by HGF has not been investigated. Nectin-based adhesions (NAs) colocalize with E-cadherin adhesions at AJs. They consist of members of the transmembrane immunoglobulin-like CAM family of the Nectins, which can engage in trans in heterotypic interactions. Their cytoplasmic tail then binds to Afadin, which links them to actomyosin and also serves as a scaffold for other junctional and actin-binding proteins. The presence of Nectins is a crucial requirement for the formation of functional cell-cell junctions in epithelial cells.Citation10 Tight Junctions (TJs) are located more apically than AJs. TJs separate apical and basolateral domains, and serve as a selective permeability barrier. TJs consist of the transmembrane adhesion receptors of the claudin family, Occludin and Junctional Adhesion Molecule (JAM) family members (JAM-A, B, C and D).Citation11-Citation13 Claudins, as well as Occludin and JAMs, can bind to members of the intracellular Zonula Occludens (ZO) family that directly link TJs to actomyosin.Citation14
The Claudin family consists of 24 different members. The expression of different combinations of Claudins determines particle size-selectivity of the diffusion barrier between epithelial cells.Citation15 Furthermore, Claudin expression levels are often found to be altered during tumorigenesis. Several tumors show decreased Claudin-7 expression at advanced stages, but many tumors also show upregulation of Claudin-3 and -4.Citation16 Claudin-1 and Claudin-3 are targets of the transcriptional repressors of the Snail family that are major determinants of EMT in tissue morphogenesis and cancer and Claudin-1 is indeed found to be low in certain breast cancers.Citation17-Citation20 The relation between Claudin expression and tumor progression is clearly complex and needs further clarification.Citation21,Citation22 HGF treatment of MDCK II cells leads to a loss of expression of Claudin-2, dependent on the activation of ERK2, but how this relates to HGFs effect on junction integrity and metastatic cell behavior is not clear.Citation23 Besides expression levels, also changes in the phosphorylation status of Claudins were found to affect their barrier function and may be involved in tumor formation. For example, phosphorylation of Threonine 192 in Claudin-3 leads to disruption of TJs in an ovarian cancer cell lineCitation24 and increased phosphorylation of its C-terminal tyrosines was found in EGFR and c-MET positive colon cancer cells.Citation25
In this study, we investigated which of the actomyosin-contacted junctional complexes is primarily affected during HGF-induced scattering. FRAP studies showed that the turnover of the TJ component Claudin-3 was greatly reduced after HGF stimulation, while other junctional proteins remained unaffected. Furthermore, we show that Claudin-3 turnover within TJs primarily stems from vesicular trafficking. We closer examined the 2 previously identified and conserved c-MET responsive tyrosine phosphorylation sites in the C-terminal tail of Claudin-3. Although we did not find a direct effect of HGF on their phosphorylation status, their mutation clearly affected the regulation of Claudin-3 trafficking by HGF as did the removal of the 2 C-terminal residues crucial for PDZ-domain interaction. In conclusion, we identified Claudin trafficking as a new target of HGF-induced signaling.
Results
AJ and TJ proteins are maintained at sites of cell-cell contact until junctions are physically disrupted during HGF-induced scattering
As outlined above, regulation of actomyosin structure by HGF affects cell-cell adhesion at multiple levels. To better understand the sequence and timing of actin reorganization and the concomitant loss of the diverse actin-connected cell-cell junction complexes, we stimulated MDCK cells with HGF and fixed and stained actomyosin and junction complexes at increasing time points after stimulation. Prior to HGF stimulation, E-cadherin is assembled in linear AJs (LAJ), that are aligned with think actin bundles (, left panel). In the first hour after HGF addition, cells spread (flatten) and the junctional actin bundles become less compact and appear to dissociate in part from the junctions. This does not lead to any obvious changes in junctional integrity ( phase 1). In the next phase, (between 1 and 3 h after HGF for most cells), increasing amounts of F-actin bundles appear in the central areas of the cells, indicating increased cytoskeletal contraction. Thin radial F-actin bundles extend to cell-cell junctions, that now remodel from LAJs into Focal AJs (FAJ), a term we recently proposed for the designation of distinct Cadherin-based adhesions physically connected to perpendicular F-actin bundles (, phase 2)Citation26. As cells progressively dissociate, internal pools of E-cadherin become visible, but E-cadherin consistently remains at the remaining sites of cell-cell contact, only to be internalized when junctions have completely dissociated (, phase 3). In previous studies we have quantified the levels of E-cadherin in remaining cell-cell contacts and found that no gradual loss of intensity occursCitation7,Citation27.
Figure 1. AJ and TJ proteins are maintained at sites of cell-cell contact until junctions are physically disrupted during HGF-induced scattering. (A) IF images of E-cadherin and actin in MDCK cells before and at increasing times after stimulation with 5 ng/µl HGF. Images are representative of distinct phases of cytoskeletal and junctional remodeling. (B, C, D) Still images from time-lapse recordings of E-cadherin-EGFP (B), Claudin-3-EGFP (C), or Nectin-1-EGFP (D) co-expressed with mCherry-p120 catenin, that show the similar manner and timing in which these 3 different actin-connected adhesion complexes are remodeled and disrupted. Scalebars indicate 5 μm.
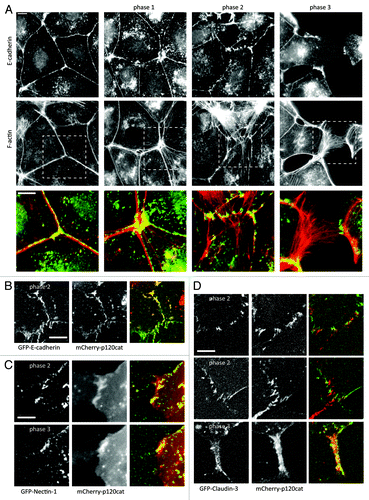
To investigate whether hierarchy exists in the dissociation of the different F-actin-connected cell-cell adhesion complexes and whether we could thus distinguish between primary and secondary targets of HGF signaling, we created MDCK cell lines stably expressing established expression constructs for E-cadherin-GFP,Citation28 GFP-Nectin-1,Citation29 or GFP-Claudin-3,Citation30,Citation31 together with mCherry-p120-catenin.Citation32 As shown in and Video S1, p120-catenin and E-cadherin remain perfectly colocalized throughout the process of junction disruption. The same holds true for E-cadherin with α-catenin and β-catenin (not shown).Citation7,Citation9 As shown in and Video S2, Nectin and p120-catenin also colocalize throughout the full process of junction disruption. Due to strong photobleaching of the extracellular GFP in the Nectin construct, exact colocalization between Nectin and p120-catenin in phase 2 was impossible to discern, but we have previously shown that Nectin and E-cadherin complexes closely, but not completely, colocalize in immature phases of cell-cell adhesion.Citation33 and Video S3 show that Claudin-3 shows a very similar behavior during junction disruption. Although it remains in different adhesion structures than p120-catenin, it remodels into punctate junctions during phase 2 and remains in adhesive patches until junctions are fully disrupted. Thus we conclude that main members of AJ and TJ adhesion complexes are maintained at sites of cell-cell contact until these junctions are physically disrupted; that there are no obvious differences in their localization and regulation when cells are induced to scatter by HGF; and that from these experiments it is impossible to pinpoint primary targets of HGF signaling among the different actin-connected cell-cell adhesion complexes.
Mobility of Claudin-3 at cell-cell junctions is affected by HGF
To investigate in more detail whether HGF differentially affects the different cell-cell junction complexes, we analyzed the dynamics of key protein from each complex at cell-cell junctions by FRAP. FRAP was performed on LAJs before HGF and 2–3 h after the addition of HGF to maximize chances of capturing the early effects of HGF signaling on junction remodeling. Bleaching was performed by a 0.300 s exposure of a focused 488nm laser beam leading to a ~1um spot in which approximately 80% of the GFP signal was lost. To plot the recovery of the different proteins, we corrected the signal intensity of each spot for general bleaching, and normalized each curve between the average intensity before bleaching (set to 1) and the lowest intensity, immediately after bleaching (set to zero). We then averaged the normalized signals from all curves at each time point. The average resultant recovery curves for all different proteins before and after HGF are shown in . It is clear from these curves that HGF does not strongly affect E-cadherin or Nectin mobility. Mobility of the TJ adhesion protein Claudin-3 was however reduced by HGF as witnessed by the reduced recovery after bleaching in and the kymographs in . The mobility of the TJ protein Occludin and the TJ-to-actin linker ZO-1 were not affected (). This indicates that the effect of HGF on the dynamics of Claudin-3 does not reflect an effect on overall TJ dynamics and is likely to be the result of a signaling event that specifically affects Claudin-3 trafficking to or stability at cell-cell junctions.
Figure 2. Dynamics of Claudin-3 at cell-cell junctions are affected by HGF. (A–E) EGFP-E-cadherin (A), - Nectin (B), Claudin-3 (C), -ZO-1 (D) and –Occludin (E) were studied by FRAP. Recovery of the fluorescent signal as a fraction of the signal prior to bleaching is shown for each of these proteins before (green diamonds) and after 2–3 h (red squares) of 5 ng/μl HGF. (F) The relative recovery was calculated for each curve as the average recovery between 150 and 200 s after bleaching. These numbers were averaged for a number of curves (n) that is indicated in the bars. Error bars represent standard error and p-values were calculated by a paired student t test. (G) Still images of Claudin-3-EGFP from before bleaching and after a 4 min recovery, with the corresponding kymographs colored artificially to allow visual inspection of the recovery process.
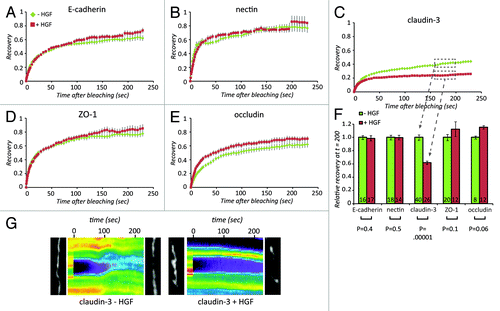
To further dissect and quantify the effects of HGF on Claudin-3 mobility, we performed single double and triple exponential curve fitting on the recovery curves from (see Fig. S1 for details). Accurate fits were only obtained using triple exponentials, indicating that more than 2 pools of Claudin-3 exist. This precluded reliable extraction of these pools and their respective dynamics from the curves. Hence we could not accurately describe the effects of HGF on a specific aspect of Claudin-3 dynamics, like for instance the loss of a rapidly recycling pool that seems obvious from . To quantify the observed differences in Claudin-3 dynamics in a systematic manner and be able to compare it directly to dynamics in manipulated situations, we plotted the average recovery attained between 150 and 200 s. These numbers are plotted in for the different adhesion proteins. Thus we conclude that Claudin-3 displays complex dynamic behavior at cell-cell junction and that the net result of HGF signaling is a reduction of its mobility, which is most likely caused by loss of a rapidly cycling pool of Claudin-3.
Claudin-3 recovers via vesicular trafficking
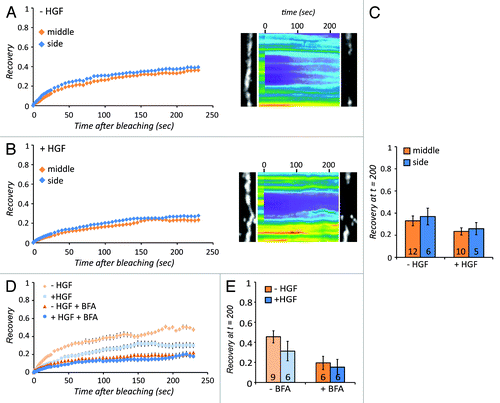
To better understand the modes of recovery that Claudin-3 proteins can undergo at the cell-cell junctions, we bleached a strip of Claudin-3-containing junctions. If lateral diffusion of Claudin-3 from adjacent free membrane would contribute, recovery would be faster at the edges of the bleached strip than in the center. As shown in , recovery was very similar in both the edges and the center of the bleached strip (), suggesting that most of the recovery does not occur by lateral diffusion within the membrane, but by delivery via vesicular trafficking from the cytosol. There might be a very minor contribution of lateral diffusion, because the side shows a slightly higher recovery than the middle (). After stimulation with HGF, the recovery of the Claudin-3 signal is still similar at the edges and at the center of the bleached strip () indicating that also in this condition, the recovering pool of Claudin-3 is delivered by vesicular trafficking. To directly test whether canonical trans-Golgi vesicular trafficking is involved in Claudin-3 recovery, we blocked antero- and retrograde trans-Golgi transport by the ARF-GEF-inhibitor Brefeldin A (BFA) (the action of BFA is extensively explained inCitation34). This resulted in a strong reduction in Claudin-3 mobility that reaches the same value in both unstimulated as well as HGF-stimulated cells (). Thus we conclude that the rapidly recovering pool of Claudin-3, of which the mobility is inhibited by HGF, is delivered at cell-cell junctions mainly through trans-Golgi vesicle trafficking.
Figure 3. Claudin-3 recovers via vesicular trafficking. (A) Recovery of Claudin-3-GFP signal in the middle vs. the side of a bleached strip of junctional Claudin-3-GFP either before or (B) after stimulation with 5 ng/µl HGF, with the corresponding kymographs and still images. Error bars represent standard error. (C) Average recovery between 150 and 200 s after bleaching. The number of recovery curves used for averaging (n) is shown in the bars. Error bars represent standard error. (D) Recovery curves showing how Claudin-3-GFP recovery is affected by treatment with the Golgi transport inhibitor BFA in both unstimulated and HGF-treated conditions. Error bars represent standard error. (E) Average recovery as in C.
Cytoskeletal remodeling is not involved in the effect of HGF on Claudin-3 turnover
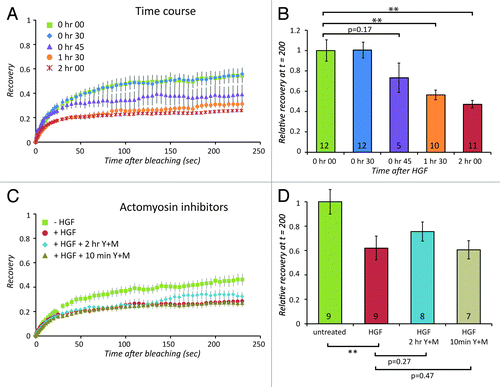
By performing FRAP measurements at increasing time points after HGF, we found that the effect of HGF on Claudin-3 dynamics only becomes apparent after 45 min and maximizes around 2 h (). This is similar in timing to the onset of phase 2 (), where the appearance of central actomyosin bundles indicates increased contraction. To investigate whether altered actomyosin organization or contractility is required for the effect of HGF on Claudin-3 turnover, we added the MLCK inhibitor ML-7 and ROCK inhibitor Y-27632 to MDCK cells stimulated with HGF. To allow the progression of the process until phase 2 and then acutely block actomyosin contractility, we added these inhibitors at 2 h after the addition of HGF, 10 min prior to the FRAP measurements (see Fig. S2A for a phospho-MLC control of inhibitor activity). The resulting recovery curve shows that this does not restore the decrease in Claudin-3 turnover that is brought about by HGF stimulation (, tan triangles). Addition of the inhibitors simultaneously with HGF was used to prevent actomyosin remodeling and phenotypic progression to phase 2 (see Fig. S2B for F-actin images that control for inhibitor activity). This only modestly affects the decrease in Claudin-3 turnover induced by HGF (, turquoise diamonds). This indicates that the effect of HGF on Claudin-3 turnover is not via a reorganization of the cytoskeleton, but a more direct result of protein modification by HGF signaling.
Figure 4. Cytoskeletal remodeling is not involved in the effect of HGF on Claudin-3 turnover. (A) Recovery of Claudin-3-EGFP signal before HGF (green squares), after 2 h of HGF (red circles), 2 h of HGF together with Y-27632 and ML-7 (blue diamonds), or 2 h of HGF with Y-27632 and ML-7 added 10 min before FRAP measurement (gray triangles) and corresponding relative recovery between 150 and 200 s after bleaching (B). The number of recovery curves used for averaging (n) is shown in the bars. (C) Timecourse of Claudin-3-EGFP recovery measured at different times after HGF stimulation: no HGF (green squares), 30 min (blue diamonds), 45 min (purples triangles), 1 h 30 min (orange circles), and 2 h (red asterisk), and corresponding relative recovery between 150 and 200 s after bleaching (D). The number of recovery curves used for averaging (n) is shown in the bars. Error bars in (A) and (C) represent standard error. ** indicates p-values smaller than or equal to 0.01 as calculated using paired student t test.
Short-term HGF signaling does not lead to a differential phosphorylation of C-terminal tyrosines in Claudin-3
Regulation of trafficking of Claudins has been attributed to interactions in their C-terminal intracellular tail.Citation30,Citation35-Citation38 Furthermore, in the tail of Claudin-3, 2 tyrosine residues were found phosphorylated in proteomics analysis of c-MET and EGFR expressing tumor cells () and their phosphorylation was reduced upon treatment with a c-MET inhibitor.Citation25 The tyrosine residue at the C-2 position is conserved among all Claudin family members commonly expressed in epithelial cells except Claudin-11 and Claudin-12, whereas the C-7 tyrosine is present only in certain members (). We took 2 approaches to investigate the importance of these sites and their phosphorylation for the HGF-induced decrease in Claudin-3 dynamics. First we isolated GFP-Claudin-3 from MDCK cells by immunoprecipitation and investigated tyrosine phosphorylation by immunoblotting and quantitative mass spectrometry analysis using stable isotope labeling by amino acids in cell culture (SILAC). As shown in , isolation of GFP-Claudin-3 was efficient and the resulting eluate was quite pure as the silverstained gel shows no major bands besides the GFP-Claudin-3 band except those that stem from marker overflow. However, by immunoblotting with anti-pY antibodies, we could not detect any pY signal in untreated or HGF-stimulated cells, whereas a strong signal was induced by incubation of the cells with the phosphatase inhibitor pervanadate (PVD) for 30 min prior to lysis. This limited amount of tyrosine phosphorylation was corroborated by our mass spectrometry analyses in which phosphorylation of tyrosines 214 or 219 was found in less than 10% of the corresponding peptides and no significant difference was found between untreated and HGF-treated cells. Thus we conclude that unlike in c-MET-overexpressing tumor cells, short-term HGF signaling does not lead to a differential phosphorylation of the C-terminal tyrosine residues in Claudin-3.
Figure 5. Short-term HGF signaling does not lead to a differential phosphorylation of C-terminal tyrosines in Claudin-3. (A) Schematic overview of several epithelially expressed Claudins. The two tyrosines indicated are involved in regulation of Claudin-3 trafficking to cell-cell junctions by HGF (B) Two tyrosines in the C-terminal tail are are conserved among species and among different Claudins. (C) Immunoprecipitation of Claudin-3-EGFP from MDCK cells labeled with either heavy or light SILAC isotopes for 3 passages. Shown are the silverstain (SS) and western blot probed with anti-GFP (WB). (D) western blot of immunoprecipitated Claudin-3-EGFP from MDCK cells, untreated or treated with HGF or pervanadate as indicated and probed with anti-GFP or anti-pY. Pervanadate was added to the lysis buffer in all samples to prevent misleading post-lysis dephosphorylation events.
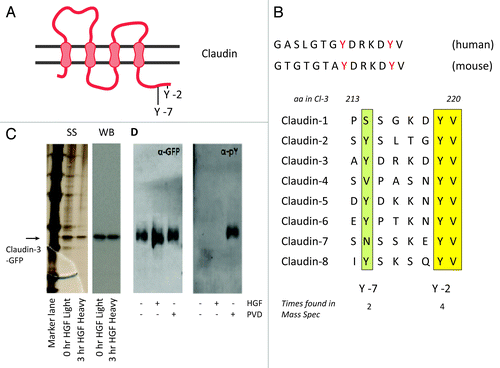
HGF-induced effects on Claudin-3 turnover depend on its C-terminal tyrosines
In a second approach to investigate their importance in HGF-induced regulation of Claudin-3 trafficking, we mutated the 2 C-terminal tyrosine residues in Claudin-3 to directly examine the importance of these sites for Claudin-3 turnover. Initially, we replaced the two tyrosines with a non-charged, non-bulky (alanine, A) residue or a phenylalanine (F) to mimic the non-phosphorylated state. Neither of these mutations affects the localization of Claudin-3 to junctions (Fig. S3) and the loss of the capacity to be phosphorylated on these residues also does not change the mobility of Claudin-3 at cell-cell junctions nor the effect of HGF thereon (). This leads to the conclusion that phosphorylation of these C-terminal tyrosines is not induced and not required for the inhibition of Claudin-3 dynamics by HGF.
Figure 6. HGF-induced effects on Claudin-3 turnover depend on its C-terminal tyrosines. (A-G) Recovery of Claudin-3-EGFP signal before HGF (green diamonds), after 2 h of HGF (red squares) for the different Claudin-3 mutants as indicated. Error bars represent standard error. (H) Relative recovery between 150 and 200 s after bleaching. The number of recovery curves used for averaging (n) is shown in the bars. P values for the difference between unstimulated and HGF-stimulated recovery values were calculated for all mutants using a paired student t test and p-values smaller than or equal to 0.01 are indicated by **.
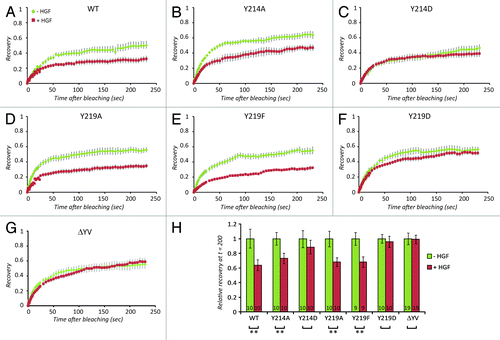
Replacing the 214 or 219 tyrosine with the charged, bulky amino acid aspartic acid (D) that is often referred to as phospho-mimetic, on the other hand completely abolished the regulation of Claudin-3 dynamics by HGF. In the Y214D mutant this appears to be caused by an HGF-comparable reduction in basal Claudin-3 dynamics in the untreated cells (), whereas in the Y219D mutant basal dynamics are unaffected, but HGF signaling has no apparent effect on this mutant (). The simplest explanation that fits these observations is that an interaction to Y214 (which is lost upon the introduction of the D residue, and possibly also upon phosphorylation of the tyrosine) is needed for basal Claudin-3 dynamics whereas an interaction to Y219 (which is lost upon introduction of a D, and possibly also upon phosphorylation of the tyrosine) is needed for the HGF effect. In Claudin-3, Y219 is part of a PDZ-domain interacting sequence (determined by the 3 C-terminal residues). To test whether an alternative disruption of this sequence similarly affects Claudin-3 regulation by HGF we deleted the 2 C-terminal residues. This had been previously shown to affect the trafficking of Claudins.Citation39 As shown in , very similar to the Y219D mutant, Claudin-3 ΔYV shows unaffected basal mobility, whereas the effect of HGF is completely abolished. Biochemical experiments to directly investigate the interaction between the Claudin-3 mutants and known PDZ-domain-containing interactors like ZO-1 failed as we could not solubilize the junction-associated pool of Claudin-3 to detect any of these interactions in co-immunoprecipitations.
Because HGF does not clearly affect phosphorylation of the 2 tyrosine residues and because mutation to un-phosphorylatable residues does not prevent the HGF-induced effects, it is unlikely that HGF signaling affects these interactions directly to regulate Claudin-3 dynamics. We conclude that Claudin-3 trafficking is regulated by HGF and that this depends on protein interactions to its C-terminal tyrosines. These interactions could be regulated by phosphorylation of these tyrosines, but this is likely not a direct effect of HGF signaling.
Discussion
We have investigated how the diverse actomyosin connected cell-cell adhesion complexes are affected by HGF to bring about junction remodeling and disruption. Contrary to common expectations, we found that the mobility of junctional Claudin-3 and not that of E-cadherin or Nectin were affected by HGF. We show that the mobility of a dynamic pool of Claudin-3 is affected by HGF-induced signaling and our results indicate that interactions to tyrosine containing sites in its C-terminus are needed for this. We discarded HGF-induced phosphorylation of these sites as a possible mechanism. Thus we have uncovered regulation of Claudin trafficking as a new target by which HGF signaling may regulate the organization and integrity of cell-cell adhesion.
Because of its central role in cell-cell adhesion, its frequent loss in metastatic cancer and its fame as the transcriptional target of EMT-driving transcription factors, E-cadherin has been the center of attention in many studies regarding HGF-induced regulation of cell-cell adhesion. There are multiple reports that show E-cadherin expression is altered in response to HGF, through transcription factors like Snail, Slug and TwistCitation40-Citation42 and also Claudin-3 expression was shown to be regulated by Snail.Citation17 Our results in cell linesCitation7 as well as primary colon carcinoma’s (unpublished), however, show that neither Snail expression, nor expression of E-cadherin are affected during the early phases of HGF-induced scattering in which junction remodeling and disruption occur. This argues that more rapid regulation of junctional proteins through modifications such as phosphorylation is a more likely event to be of importance in the HGF-induced remodeling of cell-cell adhesion. Moreover, there is no good a priori reason to place E-cadherin at the center of such events. In fact, our current results now also implicate Claudins as targets of HGF signaling to cell-cell junctions, because the mobility of Claudin-3, but not those of E-cadherin or Nectin were affected during the early stages of HGF-induced junction remodeling. This indicates that to understand how HGF regulates cell-cell adhesion remodeling, we need to consider more than the E-cadherin complex.
Notably, the majority of Claudin-3 is very stably associated with cell-cell junctions before and after HGF. This fraction is likely incorporated in functional TJ strands as has been shown by the correlation between TJ strand incorporation and loss of mobility for a number of Claudins (including Claudin-3) expressed in different cell lines.Citation43 The fact that only a fraction of Claudin-3 is dynamically associated with cell-cell junctions and that this fraction decreases upon HGF, indicates that Claudin delivery, rather than incorporation in TJs, or TJ stability per se is targeted by HGF at early stages. Our strip-bleach and inhibitor experiments () confirm that vesicular trafficking accounts for this rapid turnover and that this is the process affected by HGF, because recovery is similar in central and edge regions of a wide bleached position of a cell-cell junction and because BFA decreases the fraction of mobile Claudin-3 and abrogates additional effects of HGF on its mobility.
Regulation of Claudin trafficking has been previously attributed to interactions in its C-terminal tail.Citation44 Claudins interact with ZO-1 and 2 through their conserved C-terminal PDZ binding motif.Citation35 Loss of this interaction by deleting the last 2 amino acids (YV) that are present in all Claudin family members, or by depleting ZO-1 and ZO-2 from cells, was found to affect the trafficking of Claudins to TJs and their stability within TJs.Citation30,Citation35,Citation36 Our mutation of Y219 to a D would also disrupt PDZ domain interactions (including ZO-1). We did not find a clear effect of this mutation on basal Claudin-3 dynamics at junctions and we also found no effect of the ΔYV mutation on basal Claudin-3 dynamics. The HGF-induced loss of mobility, however was equally perturbed in both Y219D and ΔYV mutants. This indicates that indeed PDZ-domain interactions (including, but not limited to ZO-1) are needed for the HGF-induced reduction in Claudin-3 mobility. The fact that we found no effect on basal mobility, is in line with other studies, which report that the amino acids just upstream of the PDZ binding domain are responsible for basal trafficking of Claudins to the TJ, while the YV-motif is dispensable.Citation37,Citation38 In these studies trafficking of Claudin to junctions was perturbed which led to a decrease in junctional dynamics similar to the decreased basal turnover of our Y214D mutant.
The fact that stimulation of MDCK cells does not result in increased levels of phosphorylation of the C-terminal tyrosines is difficult to reconcile with the constitutive (and c-MET inhibitor sensitive) phosphorylation of these sites in c-MET positive colon cancer. It may indicate that these are cell line specific or indirect effects of prolonged c-MET activity. In any case, regulation of these phosphorylation sites is clearly not involved in HGF-dependent regulation of Claudin-3 trafficking as mutating the tyrosines to alanine residues did not perturb the effects of HGF. How then increased phosphorylation of these sites in tumor cells could be functionally linked to tumor progression does not become clear from our results. The exact explanation as to why the mutation of Y219 to D did prevent the effect of HGF remains elusive. It has been shown that HGF signaling leads to the phosphorylation of ZO-1,Citation45 therefore it could be speculated that, when associated to Claudin, this leads to alterations in their coordinated dynamics. In this light however it is surprising that basal trafficking is not affected by the Y219D or ΔYV mutations that are both very likely to cause loss of ZO-1 interaction. Furthermore, trafficking of ZO-1 itself does not seem to be affected by HGF signaling as judged from its identical recovery in the presence and absence of HGF.
It is important to note that the C-terminus is quite conserved among Claudin family members. For instance, Y219 is fully conserved among all epithelially expressed Claudin members and Y214 is present in many of them. Therefore, the effect of HGF here measured on Claudin-3 trafficking is likely to be the same for other Claudin members (although this was not formally tested). The redundancy of Claudins and their shared interactions and regulations makes it difficult to determine the functional effects of perturbation of these interactions. We have tested the effect of overexpression of all tyrosine mutants that we generated on HGF-induced MDCK cells scattering but did not observe any perturbation (Fig. S4). It is impossible to discern between redundancy among Claudins or lack of functional importance of the reduced Claudin trafficking in overall junction remodeling induced by HGF.
In conclusion, we have implicated the conserved C-terminal tail of Claudins in the regulation of their trafficking to cell-cell junctions by HGF. This highlights Claudins as new targets of HGF signaling among cell-cell adhesion complex proteins and underscores the importance of their further investigation for understanding the mechanisms by which HGF regulates the remodeling and integrity of cell-cell junctions in tissue morphogenesis and cancer.
Materials and Methods
Cell lines and culture
Madin Darby Canine Kidney strain II (MDCK II) cells were cultured in high glucose DMEM containing GlutaMAX (Gibco) and supplemented with 10% Fetal Bovine Serum (FBS, Sigma) and Penicillin/Streptomycin (Gibco) in standard 10 cm culture dishes. MDCK cells stably expressing Claudin-3-EGFP WT, E-cadherin-EGFP or Nectin-EGFP were generated by Amaxa nucleofection, followed by G418 (1 mg/ml) selection for at least 3 wk. Additional stable p120 catenin-mCherry expression was obtained by a second Amaxa nucleofection, followed by puromycin (1.3 µg/ml) selection for at least 3 wk. MDCK cells stably expressing Claudin-3-EGFP mutants were generated by lentiviral transduction, followed by puromycin (1.3 µg/ml) selection for at least 3 wk.
Immunocytochemistry, microscopy and image processing
For immunofluorescence, cells were plated overnight on glass coverslips coated o/n at 4 °C with 30 mg/ml collagen type I (Calf collagen I, Sigma) in scatter medium (DMEM supplemented with 0,5% FBS, L-glutamine, penicillin/streptomycin and 2% HEPES). At different time points after HGF (R&D) stimulation (5 ng/ml), cells were washed with PBS and fixed in 2% paraformaldehyde in PBS for 20 min. at RT, permeabilized with 0.4% Triton-X-100 for 5 min. and blocked in 2% BSA for 1 h. Phalloidin (Molecular Probes), primary (anti-E-cadherin clone 36, BD Biosciences) and secondary (coupled to Alexa-488, Molecular probes) antibody stainings were performed in 2% BSA for 1 h. Coverslips were mounted in Mowiol4–88/DABCO solution (Sigma). Cells were imaged using a widefield microscope (Nikon Eclipse Ti) with a 60x 1.49 NA Apo TIRF objective or a 40x 1.30 NA objective and an EMCCD camera (Luka, Andor). Images were enhanced for display with a background subtraction by rolling ball (15 pixel radius), unsharp mask (3 pixel radius, weight of 0.6), Gaussian blur (1 pixel radius) and brightness contrast adjustments in Image J (National Institutes of Health). Kymographs were made using Metamorph (Version 7.7.0.0).
DNA constructs and viral transduction
Claudin-3-EGFP phosphorylation mutants were generated by transferring Claudin-3-EGFP from the pCAGGS vectorCitation31 to the lentiviral pLV-CMV-ires-puro vector using the NdeI and HpaI restriction sites. Mutations were made by PCR using the following primers: 5′- CGG TAA ACT GCC CAC TTG GC -3′ (Fw primer used for all constructs); Y214A/D: 5′- cg ccc ggg TCA GAC GTA GTC CTT GCG -3′ (Rv), Y219F: 5′- cg ccc ggg TCA GAC Gaa GTC CTT GCG -3′ (Rv); Y219A: 5′- cg ccc ggg TCA GAC Ggc GTC CTT GCG -3′ (Rv); Y219D: 5′- cg ccc ggg TCA GAC GTc GTC CTT GCG -3′ (Rv). The PCR product was ligated into the NdeI and XmaI restriction sites to replace Claudin-3 WT. All clones were verified by sequencing. Lentiviral particles were isolated from HEK293T cells transiently transfected with third-generation packaging constructs and the lentiviral expression vectors. MDCK cells were transduced with supernatant containing lentiviral particles in the presence of 8 µg/ml polybrene overnight. To create stable cell lines, cells were selected with puromycin (1.3 µg/ml) for at least three weeks before being used in experiments.
FRAP
Cells were seeded in collagen-I coated (Sigma) Willco wells in regular growth medium. After 24 h, 1 h before FRAP measurements, medium was replaced by FRAP medium (Optimem supplemented with 0.5% FBS (Sigma) and penicillin/streptomycin). Cells were stimulated with 5 ng/ml HGF (Sigma), 3 μM ML-7, 10 μM Y-27632 as indicated. Cells were imaged on a Deltavision RT core system (Applied Precision) equipped with a 60x 1.3 NA objective, heated to 37 °C. Five images were taken before a ~1 μm spot was bleached with a 488 nm laser at maximum power for 0.300 s. Following bleaching, cells were imaged for another 4 min.
Image analyses were performed using Metamorph (Version 7.7.0.0). Regions of interest were drawn, and the mean fluorescence intensity was logged for each time point. Furthermore, the camera offset was determined in an area without any cells, and subtracted from each logged value. The mean fluorescence intensity of the whole image for each time point was logged to correct for background bleaching. Resulting values were stretched between the average intensity before bleaching (set to 1) and intensity immediately after bleaching (set to 0). Resulting intensity values for all bleached spots were pooled, and fit to three different equations (see Fig. S3 for details), or the average for each time point was calculated and plotted. To perform meaningful statistical analyses of the differences between unstimulated and HGF-stimulated conditions and among different mutants, the normalized values of individual recovery curves were averaged between 150 and 200 s post-bleaching to generate 1 number per curve. These numbers were than averaged over the nr of curves as indicated in the different figures and paired student t tests were used to assess significance of the differences between 2 conditions. The resulting p-values of these analyses are indicated in the figure, where “**” denotes a p-value equal to or smaller than 0.01.
Mass Spec
For quantitative Mass Spec experiments, MDCK cells were grown for 3 passages in standard SILAC medium (PAA Laboratories), supplemented with 10% 10kD dialyzed FBS, penicillin/streptomycin, L-Glutamine, l-proline, and K and R isotope-labeled amino acids. Cells were controlled for equal retention of isotope-labeled amino acids, and then used as described below.
Cells were seeded in collagen-I coated (Sigma) standard 10 cm cell culture dishes in regular growth medium. After 5 h, medium was replaced with scatter medium (see above). The next day, cells were stimulated with 5 ng/ml HGF in scatter medium (heavy isotope-labeled cells), vanadate treatment (100 µM pervanadate) or solely scatter medium (light isotope-labeled cells_. After three hours, cells were put on ice and washed twice in ice-cold PBS/1mM CaCl2 before lysis in ice-cold RIPA buffer (50 mM Tris pH 7.5, 1% NP40, 0.5% DOC, 150 mM NaCl, 0.1% SDS) supplemented with protease inhibitor cocktail (PIC, 1:1000, Roche) 1 mM orthovanadate, a tip of sodium fluoride (NaF) and phosphatase inhibitor (Sigma, 1:1000). After 5 min on ice, cell were scraped and centrifuged for 10 min at 4 °C at 13.200 rpm. Supernatant was added to pre-washed GFP-Trap_A beads slurry (Chromotek) and incubated for 2 h at 4 °C while rotating, then centrifuged for 2 min at 4 °C at 2.000 rpm and further washed according to the manufacturer’s instructions. Elution was performed in AMBIC/Urea buffer (8M Urea, 50 mM ammonium bicarbonate, supplemented with phosphatase inhibitor and protease inhibitor cocktail). Claudin-3-EGFP was run on a polyacrylamide gel. After silverstaining, the Claudin-3-EGFP band was isolated from the gel, subjected to trypsin digestion and analyzed on an orbitrap.
Scatter assay
4000–5000 cells were seeded in collagen-I (Sigma) coated non-tissue culture-treated polystyrene 48-well plates (Nunc) in regular growth medium. The next day, wells were completely filled with scatter medium (see above), and wells were sealed with silicon grease and a glass plate. Cells were imaged for 2 h before wells were briefly opened to add HGF (1.6 ng/ml) after which imaging continued for approximately 12 h.
| Abbreviations: | ||
| HGF | = | Hepatocyte Growth Factor |
| MDCK | = | Madin Darby Canine Kidney |
| ERK2 | = | Extracellular Signal-Regulated Kinase 2 |
| PI3K | = | Phosphatidyl Inositol 3 Kinase |
| ZA | = | Zonula Adherens |
| NA | = | Nectin-based Adhesion |
| AJ | = | Adherens Junction |
| TJ | = | Tight Junction |
| CAM | = | Cell Adhesion Molecule |
| JAM | = | Junctional Adhesion Molecule |
| ZO | = | Zonula Occludens |
| FRAP | = | Fluorescence Recovery After Photobleaching |
| LAJ | = | Linear Adherens Junction |
| FAJ | = | Focal Adherens Junction |
| BFA | = | Brefeldin A |
| F-actin | = | Filamentous actin |
| GFP | = | Green Fluorescent Protein |
| ROCK | = | Rho-associated kinase |
| SILAC | = | Stable Isotope Labeling by Amino Acids in Cell Culture |
| EMT | = | Epithelial to Mesenchymal Transition |
| PDZ | = | Post synaptic density protein (PSD95), Drosophila disc large tumor suppressor (Dlg1), and Zonula Occludens-1 protein (ZO-1) |
Additional material
Download Zip (969.2 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank all members of the De Rooij lab for discussions. We thank Adam Grieve and Catherine Rabouille for help with and discussions about inhibiting vesicle trafficking. Floor Twiss and Johan de Rooij were funded by a research grant from NWO (VIDI) to Johan de Rooij
References
- Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol 2003; 4:915 - 25; http://dx.doi.org/10.1038/nrm1261; PMID: 14685170
- Naldini L, Weidner KM, Vigna E, Gaudino G, Bardelli A, Ponzetto C, Narsimhan RP, Hartmann G, Zarnegar R, Michalopoulos GK, et al. Scatter factor and hepatocyte growth factor are indistinguishable ligands for the MET receptor. EMBO J 1991; 10:2867 - 78; PMID: 1655405
- Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol 2010; 11:834 - 48; http://dx.doi.org/10.1038/nrm3012; PMID: 21102609
- Matsumoto K, Nakamura T. Hepatocyte growth factor and the Met system as a mediator of tumor-stromal interactions. Int J Cancer 2006; 119:477 - 83; http://dx.doi.org/10.1002/ijc.21808; PMID: 16453287
- Eder JP, Vande Woude GF, Boerner SA, LoRusso PM. Novel therapeutic inhibitors of the c-Met signaling pathway in cancer. Clin Cancer Res 2009; 15:2207 - 14; http://dx.doi.org/10.1158/1078-0432.CCR-08-1306; PMID: 19318488
- Khwaja A, Lehmann K, Marte BM, Downward J. Phosphoinositide 3-kinase induces scattering and tubulogenesis in epithelial cells through a novel pathway. J Biol Chem 1998; 273:18793 - 801; http://dx.doi.org/10.1074/jbc.273.30.18793; PMID: 9668053
- de Rooij J, Kerstens A, Danuser G, Schwartz MA, Waterman-Storer CM. Integrin-dependent actomyosin contraction regulates epithelial cell scattering. J Cell Biol 2005; 171:153 - 64; http://dx.doi.org/10.1083/jcb.200506152; PMID: 16216928
- Mangold S, Wu SK, Norwood SJ, Collins BM, Hamilton NA, Thorn P, Yap AS. Hepatocyte growth factor acutely perturbs actin filament anchorage at the epithelial zonula adherens. Curr Biol 2011; 21:503 - 7; http://dx.doi.org/10.1016/j.cub.2011.02.018; PMID: 21396819
- le Duc Q, Shi Q, Blonk I, Sonnenberg A, Wang N, Leckband D, de Rooij J. Vinculin potentiates E-cadherin mechanosensing and is recruited to actin-anchored sites within adherens junctions in a myosin II-dependent manner. J Cell Biol 2010; 189:1107 - 15; http://dx.doi.org/10.1083/jcb.201001149; PMID: 20584916
- Takai Y, Ikeda W, Ogita H, Rikitake Y. The immunoglobulin-like cell adhesion molecule nectin and its associated protein afadin. Annu Rev Cell Dev Biol 2008; 24:309 - 42; http://dx.doi.org/10.1146/annurev.cellbio.24.110707.175339; PMID: 18593353
- Balda MS, Matter K. Tight junctions at a glance. J Cell Sci 2008; 121:3677 - 82; http://dx.doi.org/10.1242/jcs.023887; PMID: 18987354
- Tsukita S, Furuse M, Itoh M. Multifunctional strands in tight junctions. Nat Rev Mol Cell Biol 2001; 2:285 - 93; http://dx.doi.org/10.1038/35067088; PMID: 11283726
- Mandell KJ, Parkos CA. The JAM family of proteins. Adv Drug Deliv Rev 2005; 57:857 - 67; http://dx.doi.org/10.1016/j.addr.2005.01.005; PMID: 15820556
- Van Itallie CM, Fanning AS, Bridges A, Anderson JM. ZO-1 stabilizes the tight junction solute barrier through coupling to the perijunctional cytoskeleton. Mol Biol Cell 2009; 20:3930 - 40; http://dx.doi.org/10.1091/mbc.E09-04-0320; PMID: 19605556
- Anderson JM, Van Itallie CM. Physiology and function of the tight junction. Cold Spring Harb Perspect Biol 2009; 1:a002584; http://dx.doi.org/10.1101/cshperspect.a002584; PMID: 20066090
- Singh AB, Sharma A, Dhawan P. Claudin family of proteins and cancer: an overview. J Oncol 2010; 2010:541957; http://dx.doi.org/10.1155/2010/541957; PMID: 20671913
- Grotegut S, von Schweinitz D, Christofori G, Lehembre F. Hepatocyte growth factor induces cell scattering through MAPK/Egr-1-mediated upregulation of Snail. EMBO J 2006; 25:3534 - 45; http://dx.doi.org/10.1038/sj.emboj.7601213; PMID: 16858414
- Ikenouchi J, Matsuda M, Furuse M, Tsukita S. Regulation of tight junctions during the epithelium-mesenchyme transition: direct repression of the gene expression of claudins/occludin by Snail. J Cell Sci 2003; 116:1959 - 67; http://dx.doi.org/10.1242/jcs.00389; PMID: 12668723
- Ohkubo T, Ozawa M. The transcription factor Snail downregulates the tight junction components independently of E-cadherin downregulation. J Cell Sci 2004; 117:1675 - 85; http://dx.doi.org/10.1242/jcs.01004; PMID: 15075229
- Hoevel T, Macek R, Mundigl O, Swisshelm K, Kubbies M. Expression and targeting of the tight junction protein CLDN1 in CLDN1-negative human breast tumor cells. J Cell Physiol 2002; 191:60 - 8; http://dx.doi.org/10.1002/jcp.10076; PMID: 11920682
- Findley MK, Koval M. Regulation and roles for claudin-family tight junction proteins. IUBMB Life 2009; 61:431 - 7; http://dx.doi.org/10.1002/iub.175; PMID: 19319969
- Turksen K, Troy TC. Junctions gone bad: claudins and loss of the barrier in cancer. Biochim Biophys Acta 2011; 1816:73 - 9; PMID: 21515339
- Lipschutz JH, Li S, Arisco A, Balkovetz DF. Extracellular signal-regulated kinases 1/2 control claudin-2 expression in Madin-Darby canine kidney strain I and II cells. J Biol Chem 2005; 280:3780 - 8; http://dx.doi.org/10.1074/jbc.M408122200; PMID: 15569684
- D’Souza T, Agarwal R, Morin PJ. Phosphorylation of claudin-3 at threonine 192 by cAMP-dependent protein kinase regulates tight junction barrier function in ovarian cancer cells. J Biol Chem 2005; 280:26233 - 40; http://dx.doi.org/10.1074/jbc.M502003200; PMID: 15905176
- Guo A, Villén J, Kornhauser J, Lee KA, Stokes MP, Rikova K, Possemato A, Nardone J, Innocenti G, Wetzel R, et al. Signaling networks assembled by oncogenic EGFR and c-Met. Proc Natl Acad Sci U S A 2008; 105:692 - 7; http://dx.doi.org/10.1073/pnas.0707270105; PMID: 18180459
- Huveneers S, de Rooij J. Mechanosensitive systems at the cadherin-F-actin interface. J Cell Sci 2013; 126:403 - 13; http://dx.doi.org/10.1242/jcs.109447; PMID: 23524998
- Loerke D, le Duc Q, Blonk I, Kerstens A, Spanjaard E, Machacek M, Danuser G, de Rooij J. Quantitative imaging of epithelial cell scattering identifies specific inhibitors of cell motility and cell-cell dissociation. Sci Signal 2012; 5:rs5; http://dx.doi.org/10.1126/scisignal.2002677; PMID: 22763340
- Adams CL, Chen YT, Smith SJ, Nelson WJ. Mechanisms of epithelial cell-cell adhesion and cell compaction revealed by high-resolution tracking of E-cadherin-green fluorescent protein. J Cell Biol 1998; 142:1105 - 19; http://dx.doi.org/10.1083/jcb.142.4.1105; PMID: 9722621
- Krummenacher C, Baribaud I, Eisenberg RJ, Cohen GH. Cellular localization of nectin-1 and glycoprotein D during herpes simplex virus infection. J Virol 2003; 77:8985 - 99; http://dx.doi.org/10.1128/JVI.77.16.8985-8999.2003; PMID: 12885915
- Shen L, Turner JR. Actin depolymerization disrupts tight junctions via caveolae-mediated endocytosis. Mol Biol Cell 2005; 16:3919 - 36; http://dx.doi.org/10.1091/mbc.E04-12-1089; PMID: 15958494
- Matsuda M, Kubo A, Furuse M, Tsukita S. A peculiar internalization of claudins, tight junction-specific adhesion molecules, during the intercellular movement of epithelial cells. J Cell Sci 2004; 117:1247 - 57; http://dx.doi.org/10.1242/jcs.00972; PMID: 14996944
- Noren NK, Liu BP, Burridge K, Kreft B. p120 catenin regulates the actin cytoskeleton via Rho family GTPases. J Cell Biol 2000; 150:567 - 80; http://dx.doi.org/10.1083/jcb.150.3.567; PMID: 10931868
- Twiss F, Le Duc Q, Van Der Horst S, Tabdili H, Van Der Krogt G, Wang N, Rehmann H, Huveneers S, Leckband DE, De Rooij J. Vinculin-dependent Cadherin mechanosensing regulates efficient epithelial barrier formation. Biol Open 2012; 1:1128 - 40; http://dx.doi.org/10.1242/bio.20122428; PMID: 23213393
- Grieve AG, Rabouille C. Golgi bypass: skirting around the heart of classical secretion. Cold Spring Harb Perspect Biol 2011; 3:3; http://dx.doi.org/10.1101/cshperspect.a005298; PMID: 21441587
- Itoh M, Furuse M, Morita K, Kubota K, Saitou M, Tsukita S. Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with the COOH termini of claudins. J Cell Biol 1999; 147:1351 - 63; http://dx.doi.org/10.1083/jcb.147.6.1351; PMID: 10601346
- Umeda K, Ikenouchi J, Katahira-Tayama S, Furuse K, Sasaki H, Nakayama M, Matsui T, Tsukita S, Furuse M, Tsukita S. ZO-1 and ZO-2 independently determine where claudins are polymerized in tight-junction strand formation. Cell 2006; 126:741 - 54; http://dx.doi.org/10.1016/j.cell.2006.06.043; PMID: 16923393
- Rüffer C, Gerke V. The C-terminal cytoplasmic tail of claudins 1 and 5 but not its PDZ-binding motif is required for apical localization at epithelial and endothelial tight junctions. Eur J Cell Biol 2004; 83:135 - 44; http://dx.doi.org/10.1078/0171-9335-00366; PMID: 15260435
- Van Itallie CM, Colegio OR, Anderson JM. The cytoplasmic tails of claudins can influence tight junction barrier properties through effects on protein stability. J Membr Biol 2004; 199:29 - 38; http://dx.doi.org/10.1007/s00232-004-0673-z; PMID: 15366421
- Shen L, Weber CR, Turner JR. The tight junction protein complex undergoes rapid and continuous molecular remodeling at steady state. J Cell Biol 2008; 181:683 - 95; http://dx.doi.org/10.1083/jcb.200711165; PMID: 18474622
- Batlle E, Sancho E, Francí C, Domínguez D, Monfar M, Baulida J, García De Herreros A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol 2000; 2:84 - 9; http://dx.doi.org/10.1038/35000034; PMID: 10655587
- Hajra KM, Chen DY, Fearon ER. The SLUG zinc-finger protein represses E-cadherin in breast cancer. Cancer Res 2002; 62:1613 - 8; PMID: 11912130
- Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004; 117:927 - 39; http://dx.doi.org/10.1016/j.cell.2004.06.006; PMID: 15210113
- Yamazaki Y, Tokumasu R, Kimura H, Tsukita S. Role of claudin species-specific dynamics in reconstitution and remodeling of the zonula occludens. Mol Biol Cell 2011; 22:1495 - 504; http://dx.doi.org/10.1091/mbc.E10-12-1003; PMID: 21372174
- Sasaki H, Matsui C, Furuse K, Mimori-Kiyosue Y, Furuse M, Tsukita S. Dynamic behavior of paired claudin strands within apposing plasma membranes. Proc Natl Acad Sci U S A 2003; 100:3971 - 6; http://dx.doi.org/10.1073/pnas.0630649100; PMID: 12651952
- Hollande F, Blanc EM, Bali JP, Whitehead RH, Pelegrin A, Baldwin GS, Choquet A. HGF regulates tight junctions in new nontumorigenic gastric epithelial cell line. Am J Physiol Gastrointest Liver Physiol 2001; 280:G910 - 21; PMID: 11292600