Abstract
Non-coding transcripts originating from bidirectional promoters have been reported in a wide range of organisms. In yeast, these divergent transcripts can be subdivided into two classes. Some are designated Cryptic Unstable Transcripts (CUTs) because they are terminated by the Nrd1-Nab3-Sen1 pathway and then rapidly degraded by the nuclear exosome. This is the same processing pathway used by yeast snoRNAs. Whereas CUTs are only easily observed in cells lacking the Rrp6 or Rrp47 subunits of the nuclear exosome, Stable Uncharacterized Transcripts (SUTs) are present even in wild-type cells. Here we show that SUTs are partially susceptible to the nuclear exosome, but are primarily degraded by cytoplasmic 5’ to 3’ degradation and nonsense-mediated decay (NMD). Therefore, SUTs may be processed more similar to mRNAs. Surprisingly, both CUTs and SUTs were found to produce 3' extended species that were also subject to cytoplasmic degradation. The functions, if any, of these extended CUTs and SUTs are unknown, but their discovery suggests that yeasts generate transcripts reminiscent of long noncoding RNAs found in higher eukaryotes.
Introduction
RNA Polymerase II (Pol II) transcribes protein-coding genes and also generates large numbers of non-coding transcripts with mostly unknown functions. As much as 93% of the human genome may be transcribed, the majority of which is non-coding.Citation1,Citation2 In higher eukaryotes such as mammals and plants, non-coding transcripts can be just as long as protein coding transcripts and subjected to a range of transcript maturation events such as splicing and alternative 3′ end processing.Citation1,Citation3,Citation4 Some of these transcripts may feed into small RNA generating pathways to regulate gene expression via RNAi mechanisms.Citation5 Although S. cerevisiae lacks RNAi components, its genome produces many non-coding transcripts that affect the expression of protein coding genes by a variety of mechanisms.Citation4,Citation6 Examples of RNAi-independent regulation by non-coding transcription include recruitment of repressive chromatin complexes,Citation7–Citation9 modulation of protein coding gene expression by transcriptional interference,Citation10,Citation11 or activation of gene expression through nucleosome remodeling.Citation12 The mechanistic diversity of the few yeast non-coding transcripts characterized in depth to date suggests that their contribution to gene regulation is likely to be complex and dependent on the chromosomal context. It appears RNAthat multiple distinct non-coding transcript sub-species remain to be more clearly defined.
A common feature of non-coding transcripts is their low abundance, which has likely prevented their earlier entry into the spotlight.Citation13 In S. cerevisiae, one class of non-coding transcripts produced by Pol II are the “cryptic unstable transcripts” (CUTs). These are generally short (median length 440 basesCitation14) and terminated by the Nrd1-Nab3-Sen1 early termination mechanism.Citation15–Citation17 The Nrd1-Nab3-Sen1 complex also recruits the nuclear exosome to promote rapid and efficient CUT degradation, as revealed by a large increase in CUT levels upon deletion of the gene for the nuclear exosome subunit Rrp6.Citation17–Citation19 The nuclear exosome is also important for turning over ncRNAs in higher eukaryotes,Citation20,Citation21 suggesting there could be conserved mechanisms for exosome-dependent degradation of short non-coding transcripts.Citation22 However, a few CUTs are reported to be degraded by other RNA decay pathways.Citation16,Citation23,Citation24 For example, the yeast non-coding transcript SRG1 and the Ty1 transposon-associated RTL transcript are stabilized in cytoplasmic RNA decay mutants.Citation23,Citation24 Consistent with cytoplasmic export of some non-coding transcripts, association of certain CUTs with polyribosomes has also been reported.Citation23 Therefore, a proportion of intergenic CUT transcripts may evade nuclear surveillance pathways and reach the cytoplasm. More recently, another class of short non-coding transcripts, the so called “stable uncharacterized transcripts” (SUTs), have been distinguished from CUTs by their detectable levels even in cells containing a functional nuclear exosome.Citation14,Citation25 SUT steady state abundance in wild-type strains fluctuates depending on the environmental conditions, but relatively little is known about how SUT levels are regulated.Citation14
Like mRNAs, transcription of non-coding RNAs is often controlled by chromatin regulation. Accumulation of the PHO84 non-coding antisense transcript requires the histone H3K4 methyltransferase Set1 and CUTs originating from the non-transcribed spacer (NTS) regions of ribosomal DNA are elevated in strains mutant for the histone deacetylase Sir2.Citation17,Citation26 In addition, the Isw2 and Swr1 nucleosome-remodeling complexes suppress non-coding antisense expression in S. cerevisiaeCitation27 and S. pombe.Citation28 Underscoring the influence of the chromatin landscape on non-coding transcription, initiation sites of CUTs and SUTs in S. cerevisiae strongly correlate with nucleosome free regions (NFRs) located predominantly 5′ and 3′ to coding regions.Citation14 Indeed, eukaryotic promoters represent a rich source of non-coding transcripts across a range of species.Citation14,Citation20,Citation21,Citation29–Citation32 In many cases, the promoter for a protein-coding gene generates a divergent non-coding transcript from the upstream region of the promoter NFR. Promoter-associated non-coding transcripts in S. cerevisiae include both SUTs and CUTs. Although both classes arise from similar promoter configurations, we show here that SUTs are only partially susceptible to Rrp6-dependent degradation and are instead primarily affected by cytoplasmic RNA decay pathways that include NMD and Xrn1-dependent 5′ to 3′ degradation. Northern blotting also reveals longer CUT and SUT RNA species (eCUTs and eSUTs) arising from 3′ extensions of the same bidirectional promoters.
Results
Post-transcriptional suppression of CUT and SUT RNAs.
To begin exploring differentiating features between CUTs and SUTs that originate from a similar genomic configuration, four members of each class were identified for analysis. Each ncRNA originates as a divergent transcript from an annotated gene promoter and there are no known overlapping transcripts that may hinder analysis ( and Sup. Fig. 1).Citation14 Strand-specific northern blotting was used to analyze the non-coding transcripts in various mutant backgrounds. We tested deletions of several chromatin-related factors previously reported to affect some non-coding transcripts: the H3K4 methyltransferase Set1 (set1Δ); Swr1, a component of the SWR nucleosome remodeling complex for incorporating H2A.Z (swr1Δ); and the histone deacetylase Hda2 (hda2Δ). None of the tested chromatin regulators affected steady state abundance of the analyzed CUTs and SUTs (, Sup. Fig. 2 and data not shown).
In contrast, CUTs and SUTs were affected to different extents by mutating RNA processing and degradation factors ( and Sup. Fig. 2). In agreement with earlier studies,Citation14 CUTs were not detected in RRP6 strains, but accumulated in rrp6Δ cells ( and B and Sup. Fig. 2). SUTs were detectable in RRP6 cells, but showed a partial increase in rrp6Δ cells ( and E and Sup. Fig. 2). This argues that CUTs are degraded primarily by the nuclear exosome, while SUTs are also susceptible, but only partially. Although the Nrd1-Nab3-Sen1 termination complex can recruit exosome to snoRNAs for trimming or to CUTs for degradation, a hypomorphic allele of NRD1 (nrd1Δ151–214),Citation38 did not phenocopy Rrp6 depletion. Instead, extended CUT transcripts (eCUTs, ), presumably representing transcription terminator read-through species, were observed. Interestingly, eCUT species were also seen upon loss of Xrn1 (xrn1Δ), the exonuclease component of the cytoplasmic 5′ to 3′ decay pathway.Citation39 These results suggest that the majority of CUT transcription terminates by the Nrd1/Sen1 pathway and is targeted for exosome degradation. However, even when this pathway is functioning some RNA polymerases read-through the Nrd1-dependent terminator and terminate further downstream by a Nrd1-independent mechanism. These eCUTs apparently are transported to the cytoplasm where they can be degraded by Xrn1.
Consistent with SUTs being partially susceptible to the nuclear exosome, their levels were also increased in the nrd1Δ151–214 early termination mutant. However, it is clear that a significant fraction of the SUTs bypasses the nuclear degradation pathway and is instead degraded by the cytoplasmic exonuclease Xrn1 ( and E and Sup. Fig. 2). Interestingly, extended transcripts were also observed for SUTs. We can detect two SUT species in all analyzed genotypes, a shorter version with the size predicted by transcript mappingCitation14 and an extended species (eSUT) that can extend up to several kilobases. While eSUTs were detectable in wild-type cells, they were also increased in cells lacking Xrn1 ( and E and Sup. Fig. 2). In contrast to CUT and eCUT transcripts that accumulate differentially in nrd1Δ151–214 and rrp6Δ, the levels of eSUTs correlate well with the abundance of the short SUT species. These data indicate that SUTs are targeted by both nuclear (Rrp6) and cytoplasmic RNA decay pathways (5′ to 3′ decay by Xrn1). However, either the two pathways each degrade a distinct fraction of the individual SUTs or the transcripts have relatively long half-lives, because transcripts are detected even when both pathways are functional.
The basis of the differential degradation mechanisms for CUTs and SUTs remains unclear. The Nrd1/exosome pathway for 3′ end processing is preferentially used during early transcription elongation, while the polyA pathway predominates further downstream.Citation40,Citation41 The median length of SUTs is reported to be 761 nucleotides while CUTs average 440 bases,Citation14 which might account for some of the differential decay mechanisms. However, other factors must also contribute to the differences because SUT477 () is only 250 bases long, even shorter than the 300 bases of CUT095 (). Another possible determinant could be the presence of binding sites for the early termination pathway RRM-domain RNA binding components Nrd1 and Nab3.Citation18,Citation42 However, the distribution or frequency of these sequence elements does not appear to differ strikingly between CUTs and SUTs ().
Non-coding transcription processing can be coupled to effects on the chromatin environment.Citation3,Citation5,Citation17 To confirm that increases in non-coding RNAs upon Rrp6 depletion were post-transcriptional, chromatin immunoprecipitation (ChIP) was performed for TBP and the RNA polymerase II subunit Rpb3 () at the bidirectional locus for the Ero1 mRNA and the SUT285 non-coding transcript (). As expected, TBP crosslinking was strongest in the shared promoter region, but showed no difference in rrp6Δ cells compared to the wild-type control. More importantly, Pol II is detected over the non-coding and coding transcript regions at equivalent levels in WT and rrp6Δ cells. Similar results were obtained at the UTP6/CUT095 bidirectional locus (). Finally, in both backgrounds the levels of Pol II crosslinking were comparable in both directions. These results indicate that the lower levels of non-coding RNA generated from bidirectional pairs are largely due to post-transcriptional mechanisms.
Characterization of CUT and SUT degradation pathways.
RNA decay can occur by multiple distinct pathways in S. cerevisiae.Citation39 To further assess the contribution of these pathways to limiting the abundance of divergent promoter-associated non-coding transcripts, CUT095 and SUT129 RNA was analyzed by northern blot in several additional mutant strains (). As shown in , deletion of the nuclear exosome subunit Rrp6 led to a large increase in CUT levels, as well as a partial increase in SUTs. Depleting the Rrp47 subunit of this complex (rrp47Δ) gave identical results, confirming the role of the nuclear exosome in degrading these classes of transcripts. The nuclear polyA polymerases Trf4 and Trf5 are components of the TRAMP complex, which acts as an exosome cofactor.Citation43 However, neither mutant led to accumulation of the analyzed transcripts. This could indicate that transcripts originating from bidirectional promoters are targeted to the exosome independently of TRAMP, but more likely reflects functional redundancy of Trf4 and Trf5.
The requirement for the Xrn1 nuclease in SUT degradation indicates these transcripts are exported to the cytoplasm. To confirm whether the xrn1Δ effect is due to the known cytoplasmic 5′ to 3′ decay pathway or instead represents an independent role of Xrn1, additional cytoplasmic degradation mutants were analyzed. Cytoplasmic decapping by the Dcp1/Dcp2 complex precedes 5′ to 3′ decay by Xrn1. Strikingly, dcp1Δ mutants exhibited increased levels of SUT129, further implicating the cytoplasmic 5′ to 3′ decay pathway and indicating the SUT transcripts are capped. The idea that CUTs and SUTs may be capped is further supported by previous studies examining RNAs associated with the Cap Binding Protein 20 (CBP20).Citation29 Edc1 is an enhancer of decapping,Citation39 but appears to be unnecessary for efficient degradation of SUT129. Interestingly, the same pattern of stabilization by these cytoplasmic degradation mutants was observed for the extended transcripts produced from CUT095, suggesting that CUTs that bypass the Nrd1 early termination pathway and exosome degradation instead behave more like SUTs with respect to termination and export to the cytoplasm.
Degradation of SUTs by Xrn1 raises the question of whether these transcripts associate with ribosomes, given that a significant proportion of cytoplasmic mRNA degradation is translation-mediated.Citation39 A selection of yeast intergenic transcripts was previously reported to associate with polyribosomes and to degrade by the translation-dependent nonsense mediated decay (NMD) pathway.Citation23,Citation39 A upf1Δ mutant strain, defective for NMD, was analyzed here. SUT129 clearly accumulates in this mutant, although to a lesser extent than in xrn1Δ mutants. In contrast, the extended SUT129 transcript was seen at higher levels in upf1Δ versus xrn1Δ, perhaps reflecting a difference in the ability of the short and long SUT species to load onto ribosomes. In addition, the extended CUT095 transcripts also accumulated in cells lacking Upf1, indicating these are also susceptible to NMD.
A second cytoplasmic mRNA degradation pathway begins with deadenylation by the Ccr4-NOT complex, followed by exosome-mediated degradation.Citation43 However, loss of Ccr4, the cytoplasmic exosome cofactor Ski2, or the 3′ end chaperone Lsm1 did not affect SUT or eCUT levels. This result suggests that the cytoplasmic 3′ to 5′ degradation pathway does not play a role in degradation of the assayed CUTs and SUTs. Instead, non-coding transcripts originating from bidirectional transcription appear to first be susceptible to the nuclear exosome, with the cytoplasmic 5′ to 3′ decay and NMD pathways degrading the ncRNAs that make it out of the nucleus.
Characterization of eCUTs and eSUTs.
Long non-coding transcripts have been described in many organisms and are often implicated in gene regulation, but they have not received as much attention in S. cerevisiae.Citation44 Given the compact yeast genome, long ncRNAs are more likely to be overlaid by flanking transcripts. Therefore, detection of long ncRNA could be masked by methods that do not distinguish overlapping transcripts. To characterize whether the eCUT species represent extensions at the 5′ or 3′ ends of the shorter ncRNA, northern blots were hybridized with probes on either side of the annotated CUT095. If the extended transcripts arise from upstream initiation sites they would be recognized by a 5′ probe. A probe 5′ to CUT095 (P5′) detects a transcript, but of a different size and altered response to the degradation mutants compared to eCUT095 (). The probe is strand-specific and so would not hybridize with the overlapping UTP6 mRNA transcript. We have not further characterized this transcript, but it is presumably an antisense transcript to UTP6. In contrast, a probe 3′ to CUT095 (P3′) reveals transcripts of the expected size for the eCUTs. These 3′ transcripts also increase in nrd1 and xrn1 mutants, as also seen with a probe to the body of CUT095 ( and ). These data suggest that the eCUT and eSUT species represent 3′ extensions, consistent with the involvement of the Nrd1 early termination pathway in repressing their levels. To analyze the SUT129 species, antisense oligonucleotides were hybridized to the RNA, followed by RNAse H digestion and northern blot analysis using a probe in the middle of the SUT (). An oligonucleotide upstream to the annotated SUT (5′) has no obvious effect on the SUT or eSUT, consistent with this probe being outside the transcript. Digestion in the middle of the annotated SUT (M) leads to decrease of the full length SUT and appearance of a smaller band representing the cleaved RNA. A reduction of the eSUT band was also observed as expected. Most importantly, a downstream oligo (3′) caused a decrease in the eSUT band without affecting the SUT, indicating that the eSUT is a 3′ extension of the SUT.
One possibility is that the extended non-coding transcripts result from polymerases that bypass the normal termination site and then read through to the next polyadenylation site. The Pol II transcription units 3′ to the ncRNA loci analyzed here are typically protein-coding genes (Sup. Fig. 1). We calculated the predicted size for a transcript comprising the SUT/CUT extended to the next annotated 3′ end and compared this with the experimentally determined size of the extended non-coding transcripts. In most cases there was poor agreement, suggesting that this simple model was unlikely to be true ().
To test whether the extended transcripts were dependent upon the polyadenylation process that acts at protein coding genes, RNA was analyzed in a strain carrying a temperature sensitive mutation in the Rna14 subunit of cleavage factor IA (CF IA) (). The Rna14 homolog in Arabidopsis can promote early termination of non-coding antisense transcripts, suggesting it could also act on yeast non-coding transcripts.Citation3 To help visualize any unstable species, the rna14-1 mutation was also combined with rrp6Δ.Citation15,Citation45 While eSUT129 and eSUT477 transcripts are slightly more prevalent at the permissive (25°C) than non-permissive temperature (37°C), these levels correlate with the temperature rather the rna14-1 mutation. Some reduction of SUT129 and SUT477 was seen in the mutant, particularly when rrp6Δ/rna14-1 is compared to rrp6Δ at 37°C. Therefore, while SUTs may utilize the canonical mRNA 3′ end formation machinery, inactivation of CF IA did not affect the levels of eSUTs. Further characterization of the 3′ extended non-coding transcript sequences will be necessary to understand their biogenesis. However, we conclude that these long non-coding transcripts are not simply the result of termination read-through and subsequent utilization of the next 3′ downstream mRNA termination site.
Discussion
Bidirectional promoters produce non-coding transcripts in many organisms and the mechanisms are likely to be conserved, so studies in yeast should be highly relevant to all eukaryotes.Citation46 Here we explore the differences between two subclasses of ncRNAs that arise from divergent transcription at yeast mRNA promoters. Although the term CUT was originally applied generally to small, unannotated non-coding RNAs, Xu et al.Citation14 operationally divided these into transcripts that were only apparent in an rrp6Δ background (CUTs) and those that were present even with a fully functional nuclear exosome (SUTs). In agreement, we see that CUTs are strongly stabilized in cells containing mutations of the nuclear exosome components Rrp6 or Rrp47. The Nrd1/Sen1 early termination pathway is also critical, consistent with the idea that CUTs use the same pathway as snoRNAs for their termination and processing. Interestingly, mammalian PROMPTs and plant UNTs, non-coding transcripts associated with promoters, are also degraded by the nuclear exosome and it will be interesting to see whether some yet to be defined early termination pathways target this degradation pathway to these transcripts.Citation20,Citation21
We find that the SUTs analyzed here are also degraded by the nuclear exosome, but still exist at significant levels even in the presence of wild-type Rrp6 and Rrp47. This result suggests that either there are two populations of the SUT, only one of which is intrinsically susceptible to nuclear degradation, or that the SUT is not efficiently recognized by the nuclear exosome and thereby kinetically bypasses this degradation pathway. Either possibility could be explained by inefficient use of the Nrd1/Sen1 early termination pathway, which is physically coupled to exosome recruitment. So far, no obvious correlation of fewer Nrd1 and Nab3 binding sites or longer transcript size has been connected with SUT identity, although this is also true of mRNAs. It is possible that SUTs contain stronger polyadenylation sequences, which would be consistent with the effect of the rna14-1 mutant on their levels. SUTs are clearly exported from the nucleus, as they are subject to cytoplasmic degradation mediated by a combination of NMD factors, decapping enzyme, and the Xrn1 5′ to 3′ exonuclease.Citation39 Just as mutants in nuclear exosome components revealed a hidden layer of the transcriptome,Citation14,Citation16,Citation21,Citation29 it is likely that analysis of xrn1 mutants would uncover even more cryptic transcription. Intriguingly, although both the nuclear exosome and cytoplasmic pathways target SUTs, the steady state levels of these transcripts in wild-type cells suggest they have half-lives closer to that of mRNAs.
Our results are consistent with and help explain some previous results from other labs that raised questions about the metabolism of CUTs. Like SUTs, a non-coding antisense transcript arising from the long terminal repeat of the yeast Ty1 retrotransposon (designated RTL) is polyadenylated and degraded by the Xrn1 cytoplasmic degradation pathway.Citation24 Srg1 and several other short, cryptic transcripts are also susceptible to cytoplasmic decay and several were shown to associate with ribosomes.Citation23,Citation24 Although these RNAs were referred to as CUTs at the time the papers were published, by comparison to the transcripts analyzed here they would now be more accurately classified as SUTs.
These and our results indicate that multiple sequential or overlapping degradation pathways can act upon non-coding transcripts in yeast and that the functional distinction between CUTs and SUTs is largely due to which termination and degradation pathways predominate. Plasticity in choice of 3′ end formation pathway has been previously been reported for other non-coding transcript classes.Citation47,Citation48 While CUTs use the snoRNA pathway, SUTs may more closely resemble coding mRNAs. Indeed, it is interesting to speculate that these cryptic transcripts provide raw material for evolution of new coding transcripts. Although CUTs and SUTs are usually referred to as non-coding, many contain small open reading frames that could encode peptides. Some of these transcripts have been found associated with polyribosomes and depend on translation for degradation via the NMD pathway.Citation23,Citation39 Examples of small peptides with striking biological functions include the wide range of toxic peptides found in Conus snails and more recently a regulator of Drosophila embryogenesis.Citation49,Citation50
One unexpected discovery of our RNA analysis was the existence of 3′ extended transcripts emanating from the CUTs and SUTs. While these are readily apparent by northern blotting, they would likely be obscured during RNA microarray analysis because of their lower levels and overlap with adjacent gene transcripts. In addition, polyadenylated RNA is used for tilling array analysis but eCUTs and eSUTs may be under-represented in this RNA fraction.Citation14 The eCUTs appear to arise from read-through of Nrd1-dependent termination, since their levels increase dramatically in the nrd1 mutant. At least in some cases, this also appears true of the eSUTs. The eCUTs and eSUTs levels increased in mutants in NMD and cytoplasmic 5′ to 3′ decay factors, indicating they are transported out of the nucleus and may even associate with ribosomes. These RNA species will need to be further characterized in future studies, since preliminary analysis raises questions about how they are terminated and about their 3′ end formation. Based on size, they do not appear to simply be bicistronic read-through transcripts that terminate at the next annotated polyA site. Also remaining to be explored is whether these long non-coding S. cerevisiae transcripts play a role in regulating adjacent genes, either by reading into them,Citation47 through other mechanisms that could include chromatin effectsCitation12,Citation51 or action in cis.Citation26,Citation52 Long non-coding transcripts in other organisms play important roles in a range of processes, including X-chromosome inactivation,Citation53 regulation of HOX gene expression,Citation8,Citation54 mediating p53 responses associated with cancer,Citation55 roles in S. pombe centromeric heterochromatin formationCitation5 and flowering time regulation in Arabidopsis.Citation3 Given the compact genome of S. cerevisiae, eCUTs and eSUTs have great potential to be components of gene regulatory circuits.
Methods
Yeast strains.
Genotypes of strains used in this study are listed in the Supplemental Material.
RNA isolation.
Fifty ml yeast cultures were grown to mid-log phase (OD600 ≈ 0.6) and cells were collected by centrifugation. Total RNA was isolated using the hot acidic phenol method as described in reference Citation33. Final RNA concentration was determined using a Nanodrop spectrophotometer.
Northern blotting.
Thirty to forty micograms of total RNA was separated by electrophoresis on 1.5% agarose-formaldehyde-MOPS gels. The RNA was transferred to a nylon transfer membrane (Nytran SPC, Whatman) via capillary blotting in 10X SSC for 36 h. RNA was cross-linked to the nylon membrane by UV irradiation and baking at 80°C for 2 h. Fifteen micrograms of DNA size standard (100 bp ladder, NEB) was loaded to be used for transcript size estimation. The position of the ethidium bromide stained size standards was marked on the membrane with a pencil and used as reference points. The membrane was incubated in a rotating oven at 65°C in 15 ml hybridization buffer [10 mg/ml BSA, 7% SDS, 1 mM EDTA pH 8, 30% phosphate buffer [0.684 M Na2HPO4, 0.316 M NaH2PO4)] for 30 min prior to addition of radioactive probe.
Single-stranded probes were generated by incorporation of radioactive dATP into DNA using a unidirectional thermocycling reaction. Twenty microliters reactions were assembled containing five units Taq DNA polymerase, 200 µM each of dCTP, dGTP and dTTP, 5 ng of DNA template consisting of a 100–300 bp long PCR product of the probed region, 0.4 µM primer oligonucleotide (antisense to the RNA to be detected) and 6 µl of α-32P-dATP added (3,000 Ci/mmol). The tube was placed into a PCR machine and subjected to 35 cycles of the following series: denaturation at 94°C for 30 s, annealing at an experimentally determined temperature for 20 s, and extension for 45 s at 72°C. Upon completion, 80 µl of water was added to the reaction and unincorporated nucleotides were removed using a Spin-50 gel-filtration column (Biomax Inc.) according to manufacturers' instructions.
The purified probe was denatured by incubation at 95°C for 5 min and chilled on ice. The probe was added to the membrane in 15 ml hybridization solution to an approximate activity of 0.5–2 million cpm/ml buffer and incubated overnight at 65°C in a rotating oven. The membranes were washed with low stringency wash buffer (0.1x SSC, 0.1% SDS) and, when necessary to reduce background, high stringency wash buffer (2x SSC, 0.1% SDS) at 65°C. Membranes were wrapped in Saran wrap for exposure. A phosphorimager (Fuji) was used for analysis and quantification.
RNAseH digestion of RNA.
Forty micrograms of total RNA was ethanol precipitated together with 300 ng of antisense oligonucleotide. The pellet was resuspended in 1X Hybridization mix (25 mM Tris-HCl pH 7.5, 1 mM EDTA, 50 mM NaCl) and samples heated to 65°C for 10 min. The sample was slowly cooled to 30°C and 9.5 µl 2X RNAseH buffer (40 mM Tris-HCl pH 7.5, 10 mM MgCl2, 100 mM NaCl, 2 mM DTT, 60 µg/ml BSA), 0.5 µl RNAseH (Invitrogen) and 1 µl RNasein (Promega) were added. The sample was incubated at 30°C for 1 h. One hundred eighty microliters of Stop mix (0.04 mg/ml tRNA, 20 mM EDTA, 0.5 M NaCl) were added to stop the RNAseH digestion. The RNA was ethanol precipitated and the pellet washed in 70% ethanol. The pellet was air dried, resuspended in 20 µl H2O and analyzed by northern blotting.
Chromatin immunoprecipitation.
Chromatin preparation and immunoprecipitation were performed as described in reference Citation34. Precipitated or input DNA was analyzed by qPCR using a Roche Lightcycler 480. A homemade qPCR reaction mix was used following a protocol described in reference Citation35. Quantitation calculations are based on the ΔΔcT methodCitation36,Citation37 and fold-enrichment of the specific region relative to a non-transcribed telomeric region was derived as described in reference Citation34. Triplicate biological samples were each analyzed using three technical repeats.
Abbreviations
| CUT | = | cryptic unstable transcript |
| SUT | = | stable uncharacterized transcript |
| ChIP | = | chromatin immunoprecipitation |
| Pol II | = | RNA polymerase II |
| S. cerevisiae | = | Saccharomyces cerevisiae |
| S. pombe | = | Saccharomyces pombe |
| CF IA | = | cleavage factor IA |
| nt | = | nucleotide |
| kb | = | kilo base pair |
| eCUT | = | extended cryptic unstable transcript |
| eSUT | = | extended stable uncharacterized transcript; ncRNA, non-coding RNA |
Figures and Tables
Figure 1 Characterization of CUT and SUT non-coding transcripts from bidirectional promoters. (A) Schematic representation of a bidirectional promoter. Position of a strand specific probe to the body of the CUTs/SUTs is shown. (B–E) Northern blotting of non-coding transcripts in candidate chromatin and RNA processing mutants. CUTs and SUTs and the extended transcripts (eCUTs/eSUTs) are indicated on the left side of the blots while the positions of the 500 and 1,000 nucleotide size standards are marked on the right. The membranes were stripped and re-probed to detect the ACT1 actin mRNA loading control. Quantification of non-coding transcript species abundance relative to ACT1 is given in numbers below the images, “B“ signifies that no signal above background was observed.
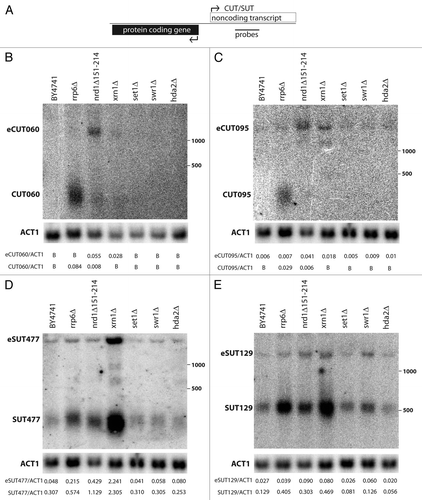
Figure 2 Stabilization of SUTs and CUTs in rrp6Δ is post-transcriptional. (A) Analysis across the bidirectional ERO1/SUT285 region. Schematic figure of the locus is at top with position of ChIP primer pairs indicated. Top graph shows ChIP signal for TBP in RRP6 (white) and rrp6Δ (gray) cells. Lower part shows ChIP for the Rpb3 subunit of RNA pol II. Data was generated by qPCR and is expressed as fold-enrichment of the indicated region relative to a non-transcribed telomeric control. Three biological and technical repeats were analyzed, error bars denote standard errors. (B) Analysis of Rpb3 across the bidirectional UTP6/CUT095 region, as in part (A).
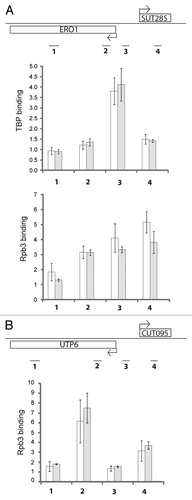
Figure 3 CUT095 and SUT129 transcript profiles in a collection of RNA decay pathway mutants. Northern blots of CUT095 (top) or SUT129 (middle) of RNA samples from the indicated RNA decay mutants are shown. The expected size transcripts and the extended eCUTs/eSUTs are indicated at left. The membranes were stripped and re-probed to detect the ACT1 (actin) mRNA as a loading control (bottom). Note that BY4741 is the isogenic “wild-type” control for strains to the left of the vertical line while W303-1B is the corresponding control for strains to the right. Quantification of non-coding transcript species abundance relative to ACT1 is given in numbers below the images, “B“ signifies that no signal above background was observed.
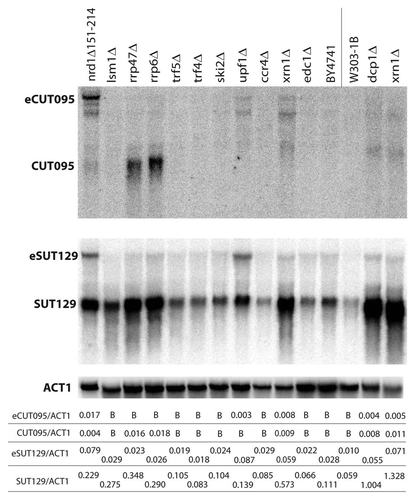
Figure 4 Characterization of 3′ extended non-coding transcripts originating from bidirectional promoters. (A) Northern blots of CUT095 were hybridized with strand-specific probes recognizing top strand transcripts as diagrammed in the schematic figure, detecting transcripts 5′ of CUT095 (P5′, left), CUT095 (P95, middle) or transcripts 3′ of CUT095 (P3′, right). Note the P95 part is the same blot that appears in . (B) Total RNA was hybridized with no oligo (-) or antisense oligos and to the regions of the SUT129 locus indicated in the diagram on top and then treated with RNAse H. The digested samples were then analyzed by northern blotting using a probe against the middle of SUT129 (gray bar). Note that the eSUT levels decrease only with the middle (M) or 3′ oligos. Quantification of the eSUT/SUT ratio of two repeats is given below, error bars denote standard errors. Note that for the M lane, the SUT signal is the combination of both smaller bands. (C) Effect of CF IA inactivation on short and extended non-coding transcripts. RNA from the indicated strains grown at permissive (25°C) or nonpermissive (37°C) temperatures were analyzed by northern blot. SUT and eSUT transcript species are indicated. The membranes were stripped and re-probed to detect the 5S rRNA (5S) as loading control (bottom). Quantification of non-coding transcript species abundance relative to 5S is given in numbers below the images, “B“ signifies that no signal above background was observed.
Table 1 Frequency of Nrd1 and Nab3 recognition sequences associated with CUTs and SUTs
Table 2 Comparison between estimated and expected non-coding transcript sizes
Additional material
Download Zip (7.4 MB)Acknowledgments
We thank Roy Parker, N. Terzi and T.S. Kim for yeast strains. S.M. is a Feodor Lynen Fellow of the Alexander von Humboldt Foundation. D.H. is an American Cancer Society Postdoctoral Fellow. This work was supported by NIH grant GM56663 to S.B.
References
- Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell 2009; 136:629 - 641
- Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007; 447:799 - 816
- Liu F, Marquardt S, Lister C, Swiezewski S, Dean C. Targeted 3′ processing of antisense transcripts triggers Arabidopsis FLC chromatin silencing. Science 2010; 327:94 - 97
- Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009; 458:223 - 227
- Grewal SI. RNAi-dependent formation of heterochromatin and its diverse functions. Curr Opin Genet Dev 2010; 20:134 - 141
- Drinnenberg IA, Weinberg DE, Xie KT, Mower JP, Wolfe KH, Fink GR, et al. RNAi in budding yeast. Science 2009; 326:544 - 550
- Camblong J, Iglesias N, Fickentscher C, Dieppois G, Stutz F. Antisense RNA stabilization induces transcriptional gene silencing via histone deacetylation in S. cerevisiae. Cell 2007; 131:706 - 717
- Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007; 129:1311 - 1323
- Houseley J, Rubbi L, Grunstein M, Tollervey D, Vogelauer M. A ncRNA modulates histone modification and mRNA induction in the yeast GAL gene cluster. Mol Cell 2008; 32:685 - 695
- Hongay CF, Grisafi PL, Galitski T, Fink GR. Antisense transcription controls cell fate in Saccharomyces cerevisiae. Cell 2006; 127:735 - 745
- Martens JA, Laprade L, Winston F. Intergenic transcription is required to repress the Saccharomyces cerevisiae SER3 gene. Nature 2004; 429:571 - 574
- Uhler JP, Hertel C, Svejstrup JQ. A role for noncoding transcription in activation of the yeast PHO5 gene. Proc Natl Acad Sci USA 2007; 104:8011 - 8016
- Mattick JS. The genetic signatures of noncoding RNAs. PLoS Genet 2009; 5:1000459
- Xu Z, Wei W, Gagneur J, Perocchi F, Clauder-Munster S, Camblong J, et al. Bidirectional promoters generate pervasive transcription in yeast. Nature 2009; 457:1033 - 1037
- Vasiljeva L, Buratowski S. Nrd1 interacts with the nuclear exosome for 3′ processing of RNA polymerase II transcripts. Mol Cell 2006; 21:239 - 248
- Wyers F, Rougemaille M, Badis G, Rousselle JC, Dufour ME, Boulay J, et al. Cryptic pol II transcripts are degraded by a nuclear quality control pathway involving a new poly(A) polymerase. Cell 2005; 121:725 - 737
- Vasiljeva L, Kim M, Terzi N, Soares LM, Buratowski S. Transcription termination and RNA degradation contribute to silencing of RNA polymerase II transcription within heterochromatin. Mol Cell 2008; 29:313 - 323
- Arigo JT, Eyler DE, Carroll KL, Corden JL. Termination of cryptic unstable transcripts is directed by yeast RNA-binding proteins Nrd1 and Nab3. Mol Cell 2006; 23:841 - 851
- Thiebaut M, Kisseleva-Romanova E, Rougemaille M, Boulay J, Libri D. Transcription termination and nuclear degradation of cryptic unstable transcripts: a role for the nrd1-nab3 pathway in genome surveillance. Mol Cell 2006; 23:853 - 864
- Preker P, Nielsen J, Kammler S, Lykke-Andersen S, Christensen MS, Mapendano CK, et al. RNA exosome depletion reveals transcription upstream of active human promoters. Science 2008; 322:1851 - 1854
- Chekanova JA, Gregory BD, Reverdatto SV, Chen H, Kumar R, Hooker T, et al. Genome-wide high-resolution mapping of exosome substrates reveals hidden features in the Arabidopsis transcriptome. Cell 2007; 131:1340 - 1353
- Steinmetz EJ, Brow DA. Repression of gene expression by an exogenous sequence element acting in concert with a heterogeneous nuclear ribonucleoprotein-like protein, Nrd1, and the putative helicase Sen1. Mol Cell Biol 1996; 16:6993 - 7003
- Thompson DM, Parker R. Cytoplasmic decay of intergenic transcripts in Saccharomyces cerevisiae. Mol Cell Biol 2007; 27:92 - 101
- Berretta J, Pinskaya M, Morillon A. A cryptic unstable transcript mediates transcriptional trans-silencing of the Ty1 retrotransposon in S. cerevisiae. Genes Dev 2008; 22:615 - 626
- Xu Z, Wei W, Gagneur J, Clauder-Munster S, Smolik M, Huber W, et al. Antisense expression increases gene expression variability and locus interdependency. Mol Syst Biol 2011; 7:468
- Camblong J, Beyrouthy N, Guffanti E, Schlaepfer G, Steinmetz LM, Stutz F. Trans-acting antisense RNAs mediate transcriptional gene cosuppression in S. cerevisiae. Genes Dev 2009; 23:1534 - 1545
- Whitehouse I, Rando OJ, Delrow J, Tsukiyama T. Chromatin remodelling at promoters suppresses antisense transcription. Nature 2007; 450:1031 - 1035
- Zofall M, Fischer T, Zhang K, Zhou M, Cui B, Veenstra TD, et al. Histone H2A.Z cooperates with RNAi and heterochromatin factors to suppress antisense RNAs. Nature 2009; 461:419 - 422
- Neil H, Malabat C, d'Aubenton-Carafa Y, Xu Z, Steinmetz LM, Jacquier A. Widespread bidirectional promoters are the major source of cryptic transcripts in yeast. Nature 2009; 457:1038 - 1042
- Seila AC, Calabrese JM, Levine SS, Yeo GW, Rahl PB, Flynn RA, et al. Divergent transcription from active promoters. Science 2008; 322:1849 - 1851
- Core LJ, Waterfall JJ, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 2008; 322:1845 - 1848
- Kapranov P, Cheng J, Dike S, Nix DA, Duttagupta R, Willingham AT, et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 2007; 316:1484 - 1488
- Keogh MC, Kurdistani SK, Morris SA, Ahn SH, Podolny V, Collins SR, et al. Cotranscriptional set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell 2005; 123:593 - 605
- Keogh MC, Buratowski S. Using chromatin immunoprecipitation to map cotranscriptional mRNA processing in Saccharomyces cerevisiae. Methods Mol Biol 2004; 257:1 - 16
- Geisberg JV, Struhl K. Analysis of protein co-occupancy by quantitative sequential chromatin immunoprecipitation. Curr Protoc Mol Biol 2005; 21:8
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 2008; 3:1101 - 1108
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001; 25:402 - 408
- Conrad NK, Wilson SM, Steinmetz EJ, Patturajan M, Brow DA, Swanson MS, et al. A yeast heterogeneous nuclear ribonucleoprotein complex associated with RNA polymerase II. Genetics 2000; 154:557 - 571
- Houseley J, Tollervey D. The many pathways of RNA degradation. Cell 2009; 136:763 - 776
- Vasiljeva L, Kim M, Mutschler H, Buratowski S, Meinhart A. The Nrd1-Nab3-Sen1 termination complex interacts with the Ser5-phosphorylated RNA polymerase II C-terminal domain. Nat Struct Mol Biol 2008; 15:795 - 804
- Gudipati RK, Villa T, Boulay J, Libri D. Phosphorylation of the RNA polymerase II C-terminal domain dictates transcription termination choice. Nat Struct Mol Biol 2008; 15:786 - 794
- Carroll KL, Pradhan DA, Granek JA, Clarke ND, Corden JL. Identification of cis elements directing termination of yeast nonpolyadenylated snoRNA transcripts. Mol Cell Biol 2004; 24:6241 - 6252
- Houseley J, Tollervey D. The nuclear RNA surveillance machinery: the link between ncRNAs and genome structure in budding yeast?. Biochim Biophys Acta 2008; 1779:239 - 246
- Koziol MJ, Rinn JL. RNA traffic control of chromatin complexes. Curr Opin Genet Dev 2010; 20:142 - 148
- Minvielle-Sebastia L, Preker PJ, Keller W. RNA14 and RNA15 proteins as components of a yeast pre-mRNA 3′-end processing factor. Science 1994; 266:1702 - 1705
- Seila AC, Core LJ, Lis JT, Sharp PA. Divergent transcription: a new feature of active promoters. Cell Cycle 2009; 8:2557 - 2564
- Steinmetz EJ, Warren CL, Kuehner JN, Panbehi B, Ansari AZ, Brow DA. Genome-wide distribution of yeast RNA polymerase II and its control by Sen1 helicase. Mol Cell 2006; 24:735 - 746
- Rondon AG, Mischo HE, Kawauchi J, Proudfoot NJ. Fail-safe transcriptional termination for protein-coding genes in S. cerevisiae. Mol Cell 2009; 36:88 - 98
- Kondo T, Plaza S, Zanet J, Benrabah E, Valenti P, Hashimoto Y, et al. Small peptides switch the transcriptional activity of Shavenbaby during Drosophila embryogenesis. Science 2010; 329:336 - 339
- Twede VD, Miljanich G, Olivera BM, Bulaj G. Neuroprotective and cardioprotective conopeptides: an emerging class of drug leads. Curr Opin Drug Discov Devel 2009; 12:231 - 239
- Hainer SJ, Pruneski JA, Mitchell RD, Monteverde RM, Martens JA. Intergenic transcription causes repression by directing nucleosome assembly. Genes Dev 2011; 25:29 - 40
- Berretta J, Morillon A. Pervasive transcription constitutes a new level of eukaryotic genome regulation. EMBO Rep 2009; 10:973 - 982
- Chow J, Heard E. X inactivation and the complexities of silencing a sex chromosome. Curr Opin Cell Biol 2009; 21:359 - 366
- Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 472:120 - 124
- Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ, Kenzelmann-Broz D, et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 2010; 142:409 - 419
- Arigo JT, Carroll KL, Ames JM, Corden JL. Regulation of yeast NRD1 expression by premature transcription termination. Mol Cell 2006; 21:641 - 651