

ABSTRACT
Quantum chemical calculations using density functional theory BP86 and M06-2X functionals in conjunction with def2-TZVPP basis sets have been carried out on the title molecules. The calculation results reveal that the N-imides R3NNX are always clearly higher in energy than the imine isomers R2NN(X)R. In the case of phosphane imides R3PNX and the isomers R2PN(X)R, the substituent R plays a critical role in determining their relative stabilities. When R is hydrogen or phenyl group, R3PNX are always higher in energy than R2PN(X)R but the former are more stable than the latter when R = Cl. Interestingly, the Me3PNX and Me2PN(X)Me are quite close in energy. The energy decomposition analysis suggests that the P–N bond in the phosphane imides R3PNX (R = H, Cl, Me, Ph; X = H, F, Cl) should be described in terms of an electron-sharing single bond between two charged fragments R3P+-NX− that is supported by (R3P)+←(NX)− π-backdonation. The π-bond contributes 14–21% of the total orbital interactions while the σ-bond provides 60–68% of ΔEorb.
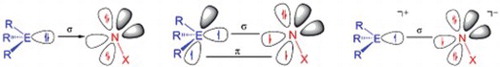
GRAPHICAL ABSTRACT
Acknowledgements
T. Y. thanks the Alexander von Humboldt Foundation for a postdoctoral fellowship. This work was supported by the Deutsche Forschungsgemeinschaft.
Disclosure statement
No potential conflict of interest was reported by the authors.
ORCID
Gernot Frenking http://orcid.org/0000-0003-1689-1197
Notes
* This paper is dedicated to the memory of Dieter Cremer.