

ABSTRACT
Density functional theory was used to compute proton affinity constants (pKa values) for the four step-wise hydrolysis reactions of the aluminum cation (Al3+ through [Al(OH)4]−). The hydrolysis species were modelled as complexes. Solvation effects were evaluated combining two approaches; initially, microsolvation of the Al-species was achieved by varying the number of explicit solvation water molecules surrounding the Al-complexes and reaction partner H2O/H3O+ species. In the second approach, the long-range polar solvent effect (global solvation) was included by using the COSMO continuum solvation model in the DFT calculations. pKa values for the hydrolysis reactions were calculated using a thermodynamic cycle, and were calculated directly from the global solvation data. The computed energies, optimised coordination structures, bond lengths, charge from natural population analyses, ionic potential, and H-bonding for the Al-hydrolysis species varied depending on the solvation approach. Similarly, calculated pKa values varied for each solvation approach. Overall cumulative pKa values calculated directly from the global solvation showed good agreement with experimental data.
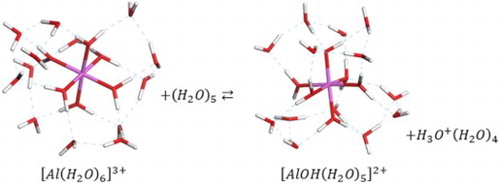
GRAPHICAL ABSTRACT
Acknowledgements
MKR acknowledges support from the National Science Foundation (CHE-1308726) for this research. We are thankful for computer time provided by the Vienna Scientific Cluster (VSC) (Project No. 70544) and the High Performance Computer Center at Texas Tech University.
Disclosure statement
No potential conflict of interest was reported by the authors.