

Abstract
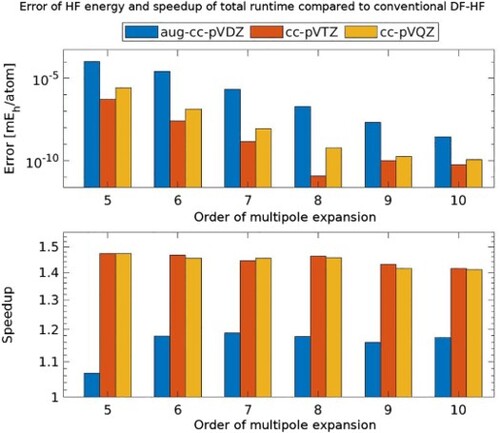
The multipole approximation is utilised for reducing the computational expenses of the exchange contribution in density fitting Hartree–Fock (DF-HF) calculations. Strategies for approximating the relevant three-centre Coulomb integrals with the multipole expansion are discussed. Based on the factorised form of the integrals, an algorithm is proposed for the evaluation of the exchange term for both conventional and local DF-HF methods. The accuracy of the resulting energies, the numerical stability of the algorithm, and the speedups achieved are benchmarked with respect to the order of the multipole expansion for various molecular systems. Our results suggest that computation times for a conventional DF-HF calculation can be reduced roughly by a factor of 1.5 for molecules of a couple of hundreds of atoms without any loss of accuracy, while the speedups are somewhat more moderate if local density fitting approximations are also deployed.
GRAPHICAL ABSTRACT
Acknowledgments
This paper is dedicated to Professor Jürgen Gauss on the happy occasion of his 60th birthday. MK wishes to express his gratitude to Jürgen for the fruitful scientific collaborations, continuous support, and friendship.
Disclosure statement
No potential conflict of interest was reported by the author(s).