

Abstract
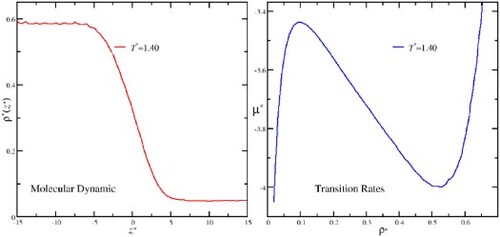
In this work, we evaluate the liquid-vapour coexistence diagram and the critical point for two different ranges, and
of the square-well dimer fluid, using two different simulation methods: (1) In the critical point vicinity, we use a new algorithm, based on transition rates, that can obtain the chemical potential as a function of the density at a given temperature and (2) Molecular Dynamics simulations using the direct coexistence technique for temperatures far below the critical region. The transition rate method has been proposed by Sastre and was used for the evaluation of the critical temperature of square-well monomers with high accuracy. The simulations in the low-temperature region were carried out using molecular dynamics simulations with the direct coexistence method and a continuous version of the square-well potential proposed recently by Zerón et al.
GRAPHICAL ABSTRACT
Acknowledgments
We also acknowledge the Centro de Supercomputación de Galicia (CESGA, Santiago de Compostela, Spain) for providing access to computing facilities. FS is very grateful with the Universidad de Huelva for its hospitality on his sabbatical leave.
Disclosure statement
No potential conflict of interest was reported by the author(s).