

Abstract
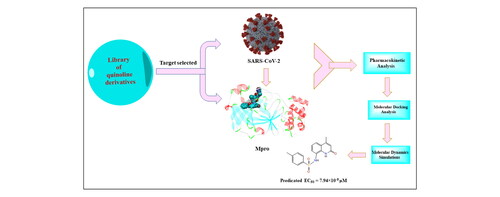
A new series of quinoline derivatives has been designed and synthesized as probable protease inhibitors (PIs) against severe acute respiratory syndrome coronavirus 2. In silico studies using DS v20.1.0.19295 software have shown that these compounds behaved as PIs while interacting at the allosteric site of target Mpro enzyme (6LU7). The designed compounds have shown promising docking results, which revealed that all compounds formed hydrogen bonds with His41, His164, Glu166, Tyr54, Asp187, and showed π-interaction with His41, the highly conserved amino acids in the target protein. Toxicity Prediction by Komputer Assisted Technology results confirmed that the compounds were found to be less toxic than the reference drug. Further, molecular dynamics simulations were performed on compound 5 and remdesivir with protease enzyme. Analysis of conformational stability, residue flexibility, compactness, hydrogen bonding, solvent accessible surface area (SASA), and binding free energy revealed comparable stability of protease:5 complex to the protease: remdesivir complex. The result of hydrogen bonding showed a large number of intermolecular hydrogen bonds formed between protein residues (Glu166 and Gln189) and ligand 5, indicating strong interaction, which validated the docking result. Further, compactness analysis, SASA and interactions like hydrogen-bonding demonstrated inhibitory properties of compound 5 similar to the existing reference drug. Thus, the designed compound 5 might act as a potential inhibitor against the protease enzyme.
Communicated by Ramaswamy H. Sarma
Quinoline derivatives have been designed as protease inhibitors against SARS-CoV-2.
The compounds were docked at the allosteric site of SARS-CoV-2-Mpro enzyme (PDB ID: 6LU7) to study the stability of protein-ligand complex.
Docking studies indicated the stable ligand-protein complexes for all designed compounds.
The Toxicity Prediction by Komputer Assisted Technology protocol in DS v20.1.0.19295 software was used to evaluate the toxicity of the designed quinoline derivatives.
Molecular dynamics studies indicated the formation of stable ligand-Mpro complexes.
Highlights
Acknowledgements
Financial assistance to Vishal K. Singh in the form of Senior Research Fellowship (Ref No: 349/CSIR-UGC NET DEC. 2017) by University Grants Commission is sincerely acknowledged. Funding by the Department of Biotechnology (India) to AS is gratefully acknowledged.
Disclosure statement
No potential conflict of interest was reported by the authors.