

Abstract
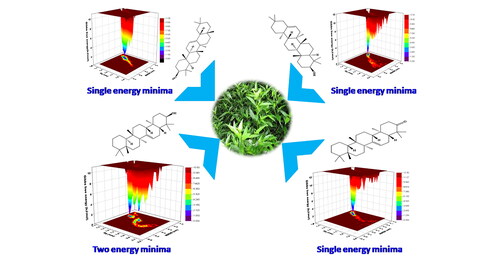
Medicinal plants the underpinning of indigenous herbal serve, are the possible source of key compounds for the development of new drugs. Hepatitis D, one of the most widespread infectious diseases associated with global public health issues. Therefore, we aim to screen natural compounds to find out potent inhibitor towards hepatitis delta antigen. Through ADMET investigation, we have screened twenty phytochemicals for this study. Additionally, using molecular docking, these phytochemicals were docked with the HDV protease which signifies the phytochemicals beta-amyrin, chiratenol, episwertenol and swertanone have a significant capability to bind with hepatitis D virus protein. The docking study was further accompanied by analyzes RMSD, RMSF, Rg, SASA, Hbond number, and principal component analysis through 100 ns MD simulations. Based on our principal component analysis, beta-amyrin, chiratenol, episwertenol and swertanone phytochemicals can be a potential drug candidates for inhibition of hepatitis D. The above observation is also supported by our Gibbs free energy landscape study. The potential therapeutic characteristics of the phytochemicals against hepatitis D inhibition offer additional support for the in vitro and in vivo studies in future.
Communicated by Ramaswamy H. Sarma
Acknowledgements
The authors are grateful to the Director, National Institute of Technology Raipur, India, for providing laboratory facility. The authors also thank the WEBGRO Macromolecular Simulations facility of University of Arkansas for Medical Sciences for providing the supercomputing facility for performing molecular dynamics simulation.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data and software availability statement
To identify the ADME and toxicity properties of phytocompounds, we used SwissADME server and DataWarrior v5.2.1.1 software. The biological activity of the studied phytocompounds were calculated using the PASS server. Molecular docking study was carried out to ascertain the binding affinity of ligand molecules and the receptor protein by using the CB-Dock2 tool. We performed simulation of the protein and its interaction with ligands by using GROMACS version 2019 with the assistance of the web server WebGro (https://simlab.uams.edu/). The details work flow is reported in the “Computational details and Methodology” section of the manuscript.