

Abstract
Saprolegnia parasitica is an oomycete responsible for a fish disease called saprolegniosis, which poses an economic and environmental burden on aquaculture production. In Saprolegnia, CHS5 of S. parasitica (SpCHS5) contains an N-terminal domain, a catalytic domain of the glycosyltransferase −2 family containing a GT-A fold, and a C-terminal transmembrane domain. No three-dimensional structure of SpCHS5 is reported yet disclosing the structural details of this protein. We have developed a structural model of full-length SpCHS5 and validated it by molecular dynamics simulation technique. From the 1 microsecond simulations, we retrieved the stable RoseTTAFold model SpCHS5 protein to explain characteristics and structural features. Furthermore, from the analysis of the movement of chitin in the protein cavity, we assumed that ARG 482, GLN 527, PHE 529, PHE 530, LEU 540, SER 541, TYR 544, ASN 634, THR 641, TYR 645, THR 641, ASN 772 residues as a main cavity lining site. In SMD analysis, we investigated the opening of the transmembrane cavity required for chitin translocation. The pulling of chitin from the internal cavity to the extracellular region was observed through steered molecular dynamics simulations. A comparison of the initial and final structures of chitin complex showed that there’s a transmembrane cavity opening in the simulations. Overall, this present work will help us understand the structural and functional basis of CHS5 and design inhibitors against SpCHS5.
Communicated by Ramaswamy H. Sarma
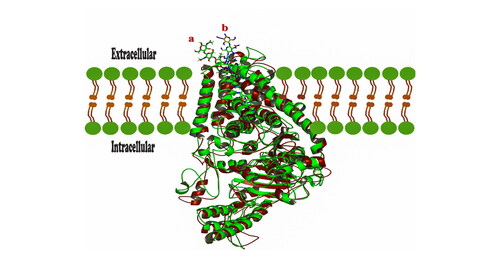
Conformational changes of chitin at the protein tunnel where ‘a’ is the orientation of chitin at 1000 ns, and ‘b’ is the initial orientation at 0 ns of the molecular dynamic simulation.
Acknowledgements
We are grateful to our donors (http://grc-bd.org/donate/) who supported building a computational platform. The authors like to acknowledge The World Academy of Science (TWAS) for purchasing High-Performance Computers for molecular dynamics simulation. We also like to give special thanks to Shafiqul Islam, Division of Infectious Diseases and Division of Computer Aided Drug Design, The Red-Green Research Centre, Dhaka, Bangladesh, for his cordial support and valuable suggestions.
Authors’ contribution
Conceptualization: Sourav Biswas, Md. Rubel Hossen, M Obayed Ullah; Investigation: Sourav Biswas, Md. Rubel Hossen, Shaila Akter, M Obayed Ullah; Data curation: Sourav Biswas, Md. Ackas Ali; Writing original draft preparation: Sourav Biswas, Md. Rubel Hossen, Shaila Akter; Writing review and editing: M Obayed Ullah; Supervision: Mohammad A. Halim, M Obayed Ullah. All authors have read and agreed to the published version of the manuscript.
Data availability
For the molecular dynamics simulations, we used the non-commercial version of Desmond (https://www.deshawresearch.com/) and for visualization (ChimeraX). We used the Desmond Incorporated Maestro Version 12.6.149, Schrödinger Suite 2020–4 and for tunnel calculation (CAVER 2.0 version). Steered molecular dynamics simulation performed using YASARA. The molecular structure of model protein, simulation output file and protein dynamic video are available from the corresponding author upon request.
Disclosure statement
The authors declare no conflict of interest regarding the publication of the manuscript. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.