

Abstract
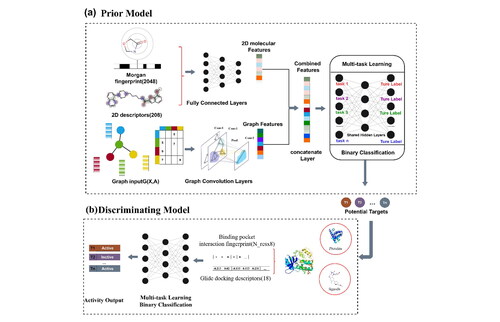
The identification of potential epigenetic targets for a known bioactive compound is essential and promising as more and more epigenetic drugs are used in cancer clinical treatment and the availability of chemogenomic data related to epigenetics increases. In this study, we introduce a novel epigenetic target identification strategy (ETI-Strategy) that integrates a multi-task graph convolutional neural network prior model and a protein-ligand interaction classification discriminating model using large-scale bioactivity data for a panel of 55 epigenetic targets. Our approach utilizes machine learning techniques to achieve an AUC value of 0.934 for the prior model and 0.830 for the discriminating model, outperforming inverse docking in predicting protein-ligand interactions. When comparing with other open-source target identification tools, it was found that only our tool was able to accurately predict all the targets corresponding to each compound. This further demonstrates the ability of our strategy to take full advantage of molecular-level information as well as protein-level information in molecular activity prediction. Our work highlights the contribution of machine learning in the identification of potential epigenetic targets and offers a novel approach for epigenetic drug discovery and development.
Communicated by Ramaswamy H. Sarma
Data availability statement
Our codes used in this study for Epigenetics target identification as well as all data involved are freely available at the GitHub website (https://github.com/CHAOJICHENG5/ETI-Strategy). Commercial databases used in this study could be accessed as follows: ChEMBL (https://www.ebi.ac.uk/chembl/), Therapeutic Target Database (http://bidd.nus.edu.sg/group/ttd/ttd.asp). Third party software packages we used are listed as follows: (1) All molecule fingerprints and physicochemical properties of molecules were calculated using the RDKit package. (2) All models were implemented based on Scikit-learn and TensorFlow and using the python programming language. (3) The protein-ligand interaction fingerprints were generated by open-source package named Open drug discovery toolkit (https://github.com/oddt/oddt). (4) We use the open source tool MolVS (https://github.com/mcs07/MolVS) to pre-process the molecules. (5) Molecular docking and complex pocket extraction were performed in Schrodinger and Molecular Operating Environment software, respectively
Disclosure statement
The authors declare no competing financial interest. We thank Dr. Huifang Li for her linguistic assistance during the preparation of this manuscript.
Authors’ contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. Lingfeng Chen and Rui Gu contributed equally to this work and should be considered as co-first authors.