

Abstract
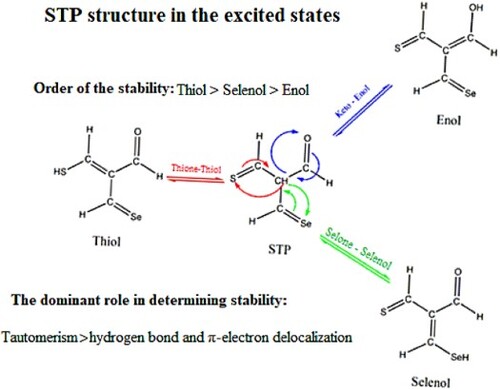
In the present study, the tautomeric process, intramolecular hydrogen bonding (IMHB), and π-electron delocalization (π-ED) of 2-selenoformyl-3-thioxo-propionaldehyde (STP) in the first singlet and triplet excited states were investigated by CIS and TD-DFT methods. The relative energies of hydrogen-bonded tautomers in both excited states indicate that the thiol/enol conformers are the most/least stable forms. In this regard, a detailed analysis of various tautomeric equilibriums, different types of hydrogen bonds, and π-electron delocalization was performed. The electronic energies of different tautomers indicate the thermodynamic preference of thiol with respect to the other forms. Furthermore, the low activation energy barriers of thione⇄thiol equilibrium also show the kinetic preference of thiol. On the other hand, the estimation of different hydrogen bond energies emphasizes the stronger IMHB of enol. Moreover, the evaluation of π-ED by the structural parameter of Gilli (λ) represents the significance of electron mobility in the enol conformers. Consequently, the duality between the IMHB and π-ED with the thermodynamic stability order of tautomers indicates that the tautomeric phenomenons play a dominant role in determining the stability of the benchmark structures in both singlet and triplet excited states.
GRAPHICAL ABSTRACT
Disclosure statement
No potential conflict of interest was reported by the author(s).