Figures & data
Figure 1 Geographical distribution of dromedaries in the United Arab Emirates infected with MERS-CoV strains that were isolated in the present study. The number of dromedaries corresponds to the number of MERS-CoV isolates from the sampling location.
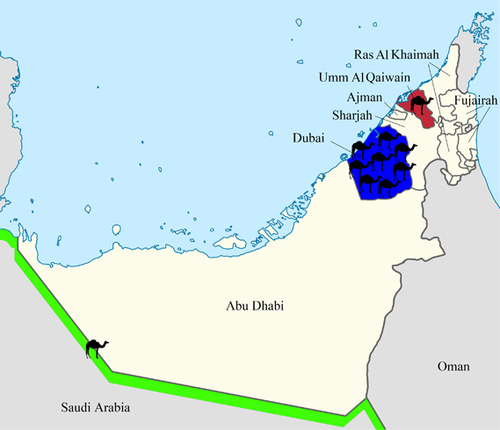
Table 1 Clinical and epidemiological data of the 10 dromedaries with MERS-CoV strains isolated and sequenced in this study
Figure 2 Maximum-likelihood phylogeny based on the complete genome sequences of 182 MERS-CoV strains. A general time-reversible model of nucleotide substitution with estimated base frequencies, the proportion of invariant sites, and the gamma distribution of rates across sites were used in the maximum-likelihood analysis. Bootstrap values are shown next to the branches. The scale bar indicates the number of nucleotide substitutions per site. MERS-CoVs from dromedaries are indicated in black circles. The ten MERS-CoV strains sequenced in the present study are colored: blue, Dubai; brown, Umm Al Quwain; green, Saudi Arabia. MERS-CoVs isolated from the United Arab Emirates are indicated by gray boxes.
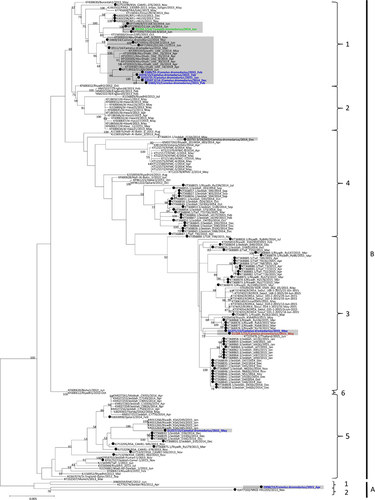
Figure 3 Maximum-likelihood phylogeny based on ORF1a, ORF1b and S gene sequences of 182 MERS-CoV strains. TIM1+I+G, GTR+I+G, and TK 2+I substitution models were selected for the ORF1a, ORF1b and S gene trees, respectively. Bootstrap values are shown next to the branches. The scale bar indicates the number of nucleotide substitutions per site. MERS-CoVs from dromedaries are indicated in black circles. The 10 MERS-CoV strains sequenced in the present study are colored: blue, Dubai; brown, Umm Al Quwain; green, Saudi Arabia. MERS-CoVs isolated from the United Arab Emirates are indicated by gray boxes.

Table 2 Comparison of amino acid substitutions between different clades and lineages
Figure 4 Detection of potential recombination events by bootscan analysis and multiple alignments. Bootscanning was conducted with Simplot version 3.5.1 (F84 model; window size, 1,500 bp; step, 200 bp) on a gapless nucleotide alignment. (A) D1271/15 and D1189.1/15 were used as the query sequences and compared with the genome sequences of a lineage 4 MERS-CoV strain Hafr-Al-Batin 1 (red, KF600628), a lineage 1 MERS-CoV strain UAE/Abu Dhabi_UAE_9 (green, KP209312) and a lineage 5 MERS-CoV strain D1157/15 (blue, KX108944). (B) D2731.3/14 was used as the query sequence and compared with the genome sequences of a lineage 1 MERS-CoV strain, FRA/UAE (green, KF745068), a lineage 4 MERS-CoV strain, KFMC-4 (red, KT121575) and a lineage 5 MERS-CoV strain, England-Qatar/2012 (blue, KC667074).
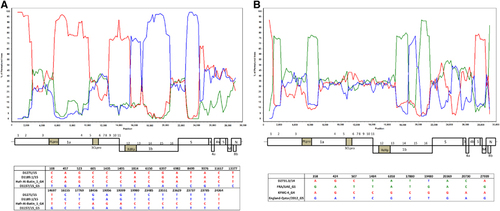
Figure 5 Maximum-likelihood trees constructed using nucleotide sequences of non-recombination regions (A), and recombination regions (B) detected using RDP4. The non-recombination region is approximately positions 1 to 14957 and 28836 to 30087, and the recombination region is approximately positions 14958 to 28835, with position numbering based on the D1189.1/15 genome sequence. Lineages are indicated by colored boxes: lineage 1, green; lineage 3, red; and lineage 5, blue.

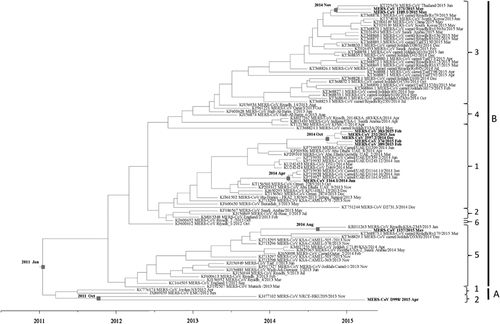
Figure 6 Estimation of time to the most recent common ancestor for MERS-CoV. The time-scaled phylogeny was summarized from all Markov chain Monte Carlo phylogenies of the concatenated coding regions (ORF1ab, S, E, M and N) of 90 phylogenetically distinct MERS-CoV genomes, which were analyzed under the relaxed-clock model with an uncorrelated lognormal distribution in BEAST version 1.8.0. The 10 MERS-CoV genomes sequenced in this study are in boldface.