
Figures & data
Table 1 The genome, fiber, hexon, and E1A sequences of adenovirus species A–G used in this study
Figure 1 Genomic organization and transcription map of HAdV-B14p1 strain GZ01. The grey arrows indicate the early, intermediate, and late transcription units; the red arrows indicate coding regions. Arrows reflect the direction of the coding transcripts.
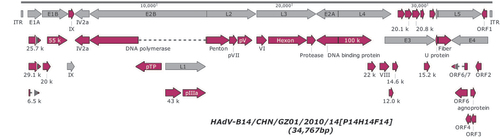
Figure 2 Identification of the genome types of HAdV-B14 strains GZ01 (Guangzhou), BJ430 (Beijing), and CHN2012 (Beijing). The in silico restriction enzyme analysis (REA) digestion patterns of HAdV-14 genomes were generated using the Vector NTI 10.3.0 software (Invitrogen Corp., San Diego, CA, USA). Genome digestion patterns of the strains are as follows: (A) de Wit or prototype (AY803294); (B) GZ01 (JQ824845); (C) 303600, reported as HAdV-B14p1 (FJ822614); (D) BJ430 (JN032132); and (E) CHN2012 (JX892927). These were analyzed with BamHI, BclI, Bgll, BglII, BstEII, EcoRI, HindIII, HpaI, SalI, SmaI, XbaI, and XhoI, as described by Li et al.Citation59 All the three recent China outbreak genomes correspond to the genome type of HAdV-14p1 and have identical REA profiles to each other. They are divergent from the HAdV-B14 prototype (1955). M: 1 kb DNA Sizing Ladder.

Figure 3 HAdV-B Inverted terminal repeats (ITR) analysis. An alignment of the left ITRs of select HAdV-B prototypes is presented, with two sets of critical sequence motifs (boxed): (1) Replication- Core origin, NF I and NF III binding sites, and (2) Sp1 and ATF binding sites. Nota bene, ‘.’ represent identical bases and ‘-’ represent deletions or gapping for sequence alignment. HAdV-B1: HAdV-B3, -B7, -B16, -B21, and -B50; HAdV-B2: HAdV-B11, -B14, -B34, -B35, and -B55.
Figure 4 Phylogenetic analysis of the E1A, fiber, and hexon genes, as well as the whole genome sequences, from all HAdV-B14 strains and representatives of other HAdV species. Nucleotide sequences of the E1A, fiber, and hexon genes, as well as limited whole genomes, are available from GenBank. Taxon names include GenBank acc. no., isolation country, strain name, and year of isolation. Phylogenetic trees were generated by using the neighbor-joining method with 1000 replicates and constructed by the MEGA 5.1.0 software (http://www.ebi.ac.uk/tools/mafft). In these analyses, default parameters were applied, with a maximum-composite-likelihood model. Bootstrap numbers shown at the nodes indicate the percentages of 1000 replications producing the clade, with values above 80 considered robust. The scale bar is in units of nucleotide substitutions per site. The China HAdV-B14 strains were indicated with filled triangles.

Figure 5 Global genome mutation map visualization of the alignment of genomes from HAdV-14 strains de Wit, 303600, BJ430, GZ01, and CHN2012. To assess the lineages and molecular evolution relationships of these pathogens, Genome Mutation Mapper (GMM; unpublished beta version) is used to provide a global visualization of the nucleotide differences, including insertions and deletions (indels). The contemporaneously circulating and independently isolated HAdV-B14p1 strains have divergent, yet similar mutation profiles, with the indels defining their lineages. For this analysis, the genome of HAdV-14p1_303600/USA/2007 is set as the reference to which the other genomes are compared. Query 1: HAdV-B14p_deWit/The Netherlands/1955 (prototype); Query 2: HAdV-B14p1_BJ430/China//2010; Query 3: HAdV-14p1_GZ01/China/2010; and Query 4: HAdV-B14p1_CHN2012/China/2012. The triangles and inverted triangles indicate deletions and insertions, respectively; blue hash lines indicate base substitutions. Select gene markers are displayed for reference (locations are approximate), as are the genome size markers noted along the bottom.
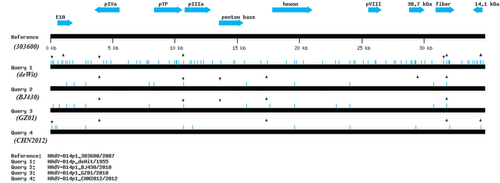
Table 2 Comparison of Mutations Across the Genomes of four HAdV-B14 Strains