

Highlights
The lowest energy conformer of the peptide backbone ring is used to design initial conformations of burkholdac C.
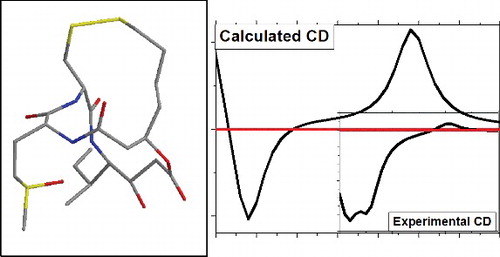
Four low energy conformers of burkholdac C are characterised by conformational geometry optimisation and electronic circular dichrosim calculations.
Taking into account the functional, basis and solvent factors, the calculated electronic circular dichrosim agrees with the experiment.
ABSTRACT
This is a theoretical investigation of conformational analysis leading to determination of the most abundant conformers of burkholdac C. For this purpose, we applied methods of molecular modelling at molecular mechanics, density functional theory and time-dependent functional theory levels. As a consequence, we determined in vacuum the pool of four lowest energy conformations. To provide correlation between theoretical and experimental signals, we compared the calculated electronic circular dichrosim spectrum with the B3LYP and CAM-B3LYP functionals to the experimental results. Taking into account the functional, basis and solvent factors, the CAM-B3LYP results are preferable
Acknowledgments
We acknowledge the important help from reviewers and editors of this journal in course of the revision and publication of this paper.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental data
Supplemental data for this article can be accessed at http://dx.doi.org/10.1080/00268976.2015.1112924.