

ABSTRACT
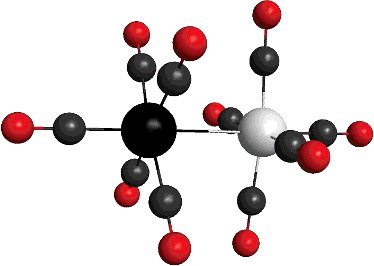
The electronic spectra of 12 isoelectronic binuclear decacarbonyls MM′(CO)10 have been calculated, using time-dependent density functional theory (TD-DFT). The geometry has been optimised using the B3LYP functional. The energy of some well-known bands is calculated with the same functional for Mn2(CO)10 and other compounds as a test, using the same functional. The calculated values for the energy and the oscillator strength are in good fitting with experimental and reference data, and they show very small dependence on the basis. Then, the same calculation has been carried out for all the compounds up to 150 states. The strongest calculated transitions are summarised and assigned.
Disclosure statement
No potential conflict of interest was reported by the author.