

ABSTRACT
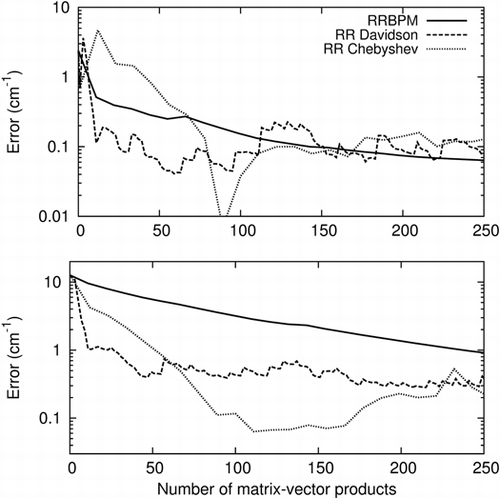
Vibrational spectra and wavefunctions of polyatomic molecules can be calculated at low memory cost using low-rank sum-of-product (SOP) decompositions to represent basis functions generated using an iterative eigensolver. Using a SOP tensor format does not determine the iterative eigensolver. The choice of the interative eigensolver is limited by the need to restrict the rank of the SOP basis functions at every stage of the calculation. We have adapted, implemented and compared different reduced-rank algorithms based on standard iterative methods (block-Davidson algorithm, Chebyshev iteration) to calculate vibrational energy levels and wavefunctions of the 12-dimensional acetonitrile molecule. The effect of using low-rank SOP basis functions on the different methods is analysed and the numerical results are compared with those obtained with the reduced rank block power method. Relative merits of the different algorithms are presented, showing that the advantage of using a more sophisticated method, although mitigated by the use of reduced-rank SOP functions, is noticeable in terms of CPU time.
Acknowledgements
Some of the calculations were done on computers purchased with a grant for the Canada Foundation for Innovation. This research was funded by the Natural Sciences and Engineering Research Council of Canada. The PMMS (Pôle Messin de Modélisation et de Simulation) is gratefully acknowledged for providing us with computer time. Tucker Carrington thanks André D. Bandrauk for his infectious and inspiring love of science.
Disclosure statement
No potential conflict of interest was reported by the authors.