

ABSTRACT
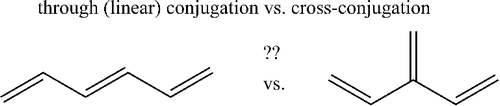
The effects of ‘through-conjugation’ (TC) vs. ‘cross-conjugation’ (CC) have been studied in a series of [n]dendralene analogues with the aid of high-level molecular orbital (MO) calculations using composite G3MP2 method. The energy differences between TC and the corresponding CC alkenes have been determined and found to be small and comparable to the conformational barrier for rotation around the C–C single bond. The destabilisation in CC vs. TC alkenes per C = C unit does not show different trends within the even and odd series of [n]dendralenes. The observed differences in UV–vis and nuclear magnetic resonance (NMR) spectra and chemical reactivities between even and odd [n]dendralenes are thus solely due to conformational preferences which of course affect C = C bond conjugation (π-delocalisation). We also propose new partially hydrogenated graphene materials (‘dendraphenes’) which were introduced in the isodesmic reactions designed to study CC.
Acknowledgments
The author thanks Charles Sturt University for the financial support of this work.
Disclosure statement
No potential conflict of interest was reported by the authors.