

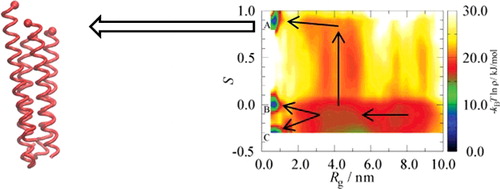
ABSTRACT
We present a simple coarse-grained model of the effective interaction for charged amino acid residues, such as Glu and Lys, in a water solvent. The free-energy profile as a function of the distance between two charged amino acid side-chain analogues in an explicit water solvent is calculated with all-atom molecular dynamics simulation and thermodynamic integration method. The calculated free-energy profile is applied to the coarse-grained potential of the effective interaction between two amino acid residues. The Langevin dynamics simulations with our coarse-grained potential are performed for association of a small protein complex, GCN4-pLI tetramer. The tetramer conformation reproduced by our coarse-grained model is similar to the X-ray crystallographic structure. We show that the effective interaction between charged amino acid residues stabilises association and orientation of protein complex. We also investigate the association pathways of GCN4-pLI tetramer.
Acknowledgment
The calculations were partly performed at the Research Center for Computational Science, Okazaki, Japan, and the Center for Computational Sciences, University of Tsukuba, Tsukuba, Japan.
Disclosure statement
No potential conflict of interest was reported by the authors.