
Figures & data
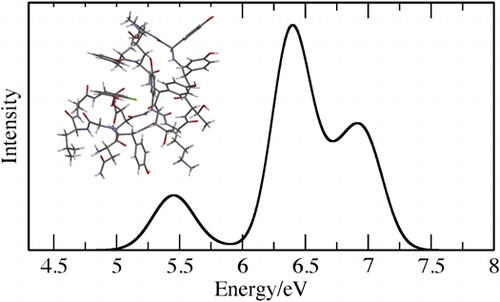
Table 1. Effect of quadrature grid relative to the (50,194) grid for TDDFT calculations for the lowest 30 states of benzene, diamide, histidine and Cr(CO)6. The 6-311+G* basis set is used, and the percentage error in the oscillator strength of the most intense transition is shown.
Table 2. Effect of truncation of the virtual subspace with the CAM-B3LYP exchange-correlation functional and 6-311+G* basis set for the lowest 30 states. The percentage error in the oscillator strength of the most intense transition is shown.
Figure 1. Variation in the computed PBE/6-31+G* spectra for (a) coronene, (b) β-carotene and (c) zinc porphyrin with the size of the virtual orbital subspace. X% indicates that excitations to the highest energy X% of virtual orbitals are excluded from the excitation space in the TDDFT calculation.
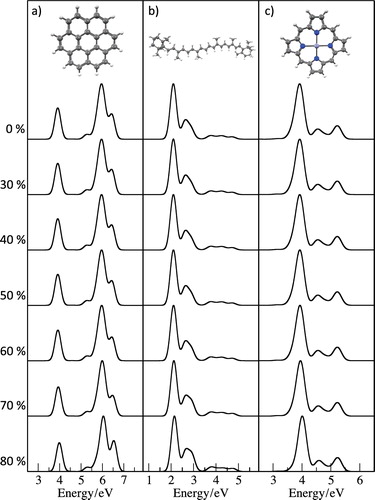
Figure 2. Molecular structures of the large molecules studied.
Figure 3. Computed spectra for (a) C60, (b) P3B2 and (c) ramoplanin. For (a) and (c) experimental data from references [Citation33,Citation34] is also shown.
![Figure 3. Computed spectra for (a) C60, (b) P3B2 and (c) ramoplanin. For (a) and (c) experimental data from references [Citation33,Citation34] is also shown.](/cms/asset/595763fb-8bc9-4e08-b442-32bdf5df06f2/tmph_a_1430388_f0003_oc.jpg)