

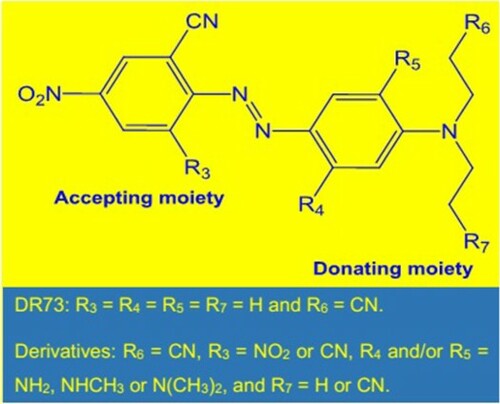
ABSTRACT
Effects of substituent groups on aqueous solubility, chemical reactivity and absorption wavelength of Disperse red 73 (DR73) were theoretically investigated using density functional theory (DFT) method. This study is in view of introducing new monoazo disperse dyes with improved aqueous solubility, absorption behaviour, and degradation tendency. Aqueous solubility was calculated using Cramer et al. solubility equation and the parameters used were obtained using conductor-like screening model for realistic solvation (COSMO-RS). Chemical reactivity and catalytic affinity were evaluated using some molecular orbital dependent reactivity descriptors. Susceptibility of the molecules to radical attacks was predicted using Fukui functions. Absorption wavelength was calculated using the B3LYP functional with the 6-31G* basis set. The results showed that electron withdrawing −CN substituent promoted aqueous solubility of DR73 more than the −NO2 counterpart, while the solubility is mostly enhanced by −NH2 substituent compared to the other electron donating groups studied. Most of the derivatives yielded longer absorption wavelength in the visible region and are chemically less stable (i.e. more degradable) compared to the parent molecule. Among the photocatalysts considered, ZnO is predicted as the most efficient candidate for degradation of the dyes. Predicted properties showed remarkable sensitivity to substitution positions in the parent molecule.
GRAPHICAL ABSTRACT
Acknowledgments
The authors would like to acknowledge the Department of Applied Chemistry-Centre of Nanomaterials Science Research (CNSR), Faculty of Science-University of Johannesburg (TTK14052167682) for providing financial aid and the Centre for High Performance Computing (CHPC, South Africa) for providing the needed computational facilities for this work.
Disclosure statement
No potential conflict of interest was reported by the authors.