

Abstract
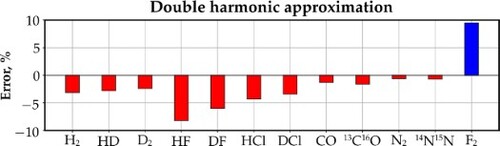
Raman intensities in molecular spectra are usually computed within double harmonic approximation. This procedure relies on treating a vibrating molecule as a collection of harmonic oscillators and on the assumption that polarisability tensor invariants display linear variations around the molecular equilibrium geometry. This methodology, originally formulated by Placzek, constitutes the theoretical foundation for computing Raman intensities in standard quantum chemistry packages. However, the two assumptions underlying double harmonic approximation have not been sufficiently tested. In this work, we employed exact anharmonic ro-vibrational wave functions and distance-dependent polarisability invariants together with their harmonic approximants to investigate the discrepancies in Raman intensities of the fundamental transitions in 12 diatomic molecules, caused by double harmonic approximation. We found that: (i) the errors in total Raman intensities were between −8.2% and +9.5%, (ii) the largest discrepancy was observed for F2, where the polarisability invariants could not be adequately modelled by their linear approximants, and (iii) quantum chemical methods fail to predict reliable polarisability invariants at non-equilibrium molecular geometries; the associated errors in Raman intensities are huge and completely overshadow the shortcomings of double harmonic approximation. We communicate here an urgent need for developing accurate methods capable of computing reliable polarisabilities also at distorted geometries.
GRAPHICAL ABSTRACT
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Online repository at GitHub [Citation72], indexed with DOI (10.5281/zenodo.6126144), contains the following auxiliary data: (i) datasets on the polarisability invariants for H2, HD, D2 (generated using the CCSD methodology) and for HF, HCl, CO, N2, and F2 (generated using the CASSCF methodology), (ii) the potential energy curves used in the computations of exact and approximate ro-vibrational wave functions, (iii) ro-vibrational wave functions and
for all the studied molecules (including isotopologues) with v = 0, 1 and J = 0, 1, 2, 3, (iv) python [Citation67,Citation80,Citation81] program implementing the collocation method to produce the ro-vibrational energy levels and wave functions for a given potential, and (v) python program using the collocation method for determination of an optimal potential energy curve reproducing a given dataset of experimental ro-vibrational energies and transition frequencies.
Notes
1 Total wave function here is expressed as product of harmonic oscillator wave functions of each normal mode, i.e. , and similarly
.
2 This approach also covers the infrared intensities where the polarisability operator is replaced by the dipole moment operator.
3 We limit our discussion to the transitions in the ground electronic state which is non-degenerate. Only off-resonance Raman scattering intensities are studied here.