

Abstract
The UV absorption spectrum of all-trans-octatetraene is reinvestigated theoretically, focussing on the strongly dipole-allowed –
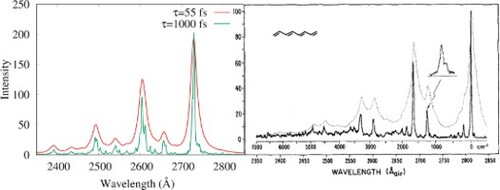
transition. The dynamical calculations rely on ab initio multistate CASPT2 (MS-CASPT2) computations of the underlying potential energy surfaces and coupling elements, and the multiconfiguration time-dependent Hartree (MCTDH) method for solving the time-dependent Schrödinger equation for the nuclear motion. The vibronic structure of the absorption band near 2700 Å is well reproduced, better than ever before by a purely ab initio approach. The C–C single bond and
double bond stretching modes dominate the vibrational structure of the transition. While the latter resembles nearly harmonic motion on weakly coupled potential energy surfaces, the time-dependent calculations reveal a subpicosecond electronic population transfer from the
state to the lower-lying
state proceeding on a time scale of
. The details of this internal conversion process and its relation to the conical intersection between the two excited states are discussed, paying particular attention to the impact of the
–
energy gap on the dynamics.
GRAPHICAL ABSTRACT
Acknowledgments
This paper is dedicated to Prof. Peter G. Szalay on the occasion of his 60th birthday. We are indebted to Prof. Andreas Dreuw for valuable discussions and a careful reading of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).