
Figures & data
Figure 1. PRISMA flow diagram of the study selection process.
Figure 2. Risk of bias graphs. Graph A depicts the assessment of the risk of selection, performance, detection, attrition and other biases using the SYRCLE’s risk of bias tool. Graph B depicts the reporting of six key quality indicators. Review author’s decision of individual items are represented as absolute numbers of included studies.
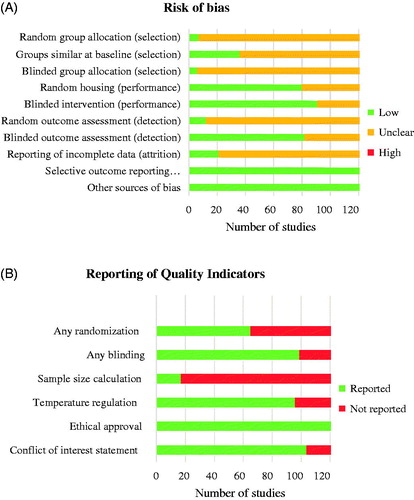
Figure 3. Schematic overview of dorsal root ganglion and spinal cord to indicate the various molecular mechanisms that contribute to paclitaxel-induced peripheral neuropathy. Paclitaxel administration to rodents induces damage to distal nerves observed as a loss of peripheral nerve fibers most pronounced at the terminal endings, thinning of myelin sheet surrounding mainly large nerve fibers, leading to reduced nerve conduction velocity and damage to neurons, as observed by an increase in ATF3, caspase-3 and TUNEL expression in the somatas of sensory neurons. Nerve damage may be caused by mitochondrial dysfunction: paclitaxel treatment results in swollen and vacuolated mitochondria with reduced ATP production. Faulty mitochondrial function and inadequate expression of several antioxidant enzymes (SOD, GPx, GSH, MDA) lead to increased intracellular ROS levels. ROS may activate the NLRP3 inflammasome in peripheral nerves and induce apoptosis. Additionally, expression and activity changes of TRP channels (e.g., TRPV1) and ion channels (e.g., Nav1.7) are observed, resulting in changes in excitability. Activation of increased translation controls may pertain to these changes in the excitability of sensory neurons. These changes are (partially) induced by mast cell degranulation and release of mediators (e.g., tryptase) that activate TLR4, further stimulating inflammatory responses, including the release of pro-inflammatory cytokines and chemokines that may affect transduction and excitability. Paclitaxel is capable of directly activating TLR4, directly stimulating an inflammatory response. Furthermore, other TLR receptors, such as TLR9, contribute to pain development in paclitaxel-treated rodents. Damaged neurons release damage-associated molecular patterns (DAMPs), which further activate TLR4 on various immune cells. Furthermore, various chemotactic cytokines secreted by damaged neurons recruit immune cells, leading to immune cell recruitment and activation in the DRG and the spinal cord. Macrophages, predominantly skewed to the pro-inflammatory M1 type, infiltrate the DRG, where they release pro-inflammatory cytokines that activate and sensitize sensory neurons. Macrophages also release HMGB1, which activates TLR4 on sensory neurons to contribute to pain development. Some reports indicate T cell recruitment to the DRG and spinal cord. T cells can produce the anti-inflammatory IL-10, which may aid in resolution of paclitaxel-induced transient neuropathy. Activated immune cells and also neurons secrete a plethora of inflammatory mediators, such as TNF, IL-17, IL-20 and CCL2, activating a variety of downstream inflammatory pathways. Within the spinal cord, paclitaxel treatment causes glial cell activation of predominantly spinal cord astrocytes, downregulation of glutamate uptake transporters on astrocytes and induction of cytokine release by astrocytes. Microglia activation is less clear. Finally, paclitaxel affects synaptic transmission regulation through upregulation of 5-HT1a receptors and neuropeptide substance P expression. Spinal excitatory synaptic transmission is also increased through σ1R and LPA receptors and dysregulated through activation of presynaptic NMDARs.
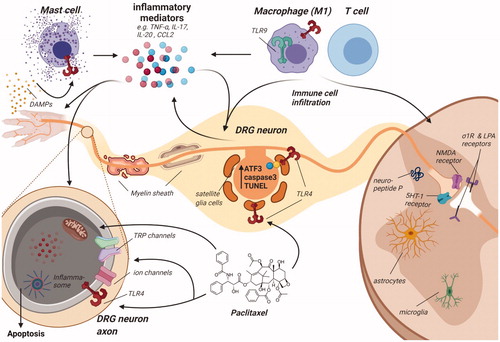
Table 1. Increased expression of TRP channels following paclitaxel treatment.
Table 2. Increased expression of voltage-gated channels following paclitaxel treatment.