

Abstract
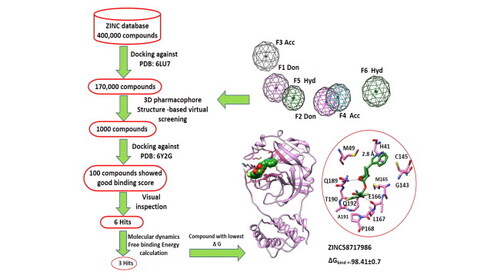
The outbreak caused by a coronavirus 2 has required quick and potential treatment strategies. The main protease enzyme Mpro plays an important role in the viral replication which renders it an important target for discovering SARS-CoV-2 inhibitors. In this study, 3D pharmacophore structure-based virtual screening and molecular docking were conducted using MOE and Bristol University Docking Engine (BUDE). Around 400,000 molecules of ZINC15 database were docked against the crystal structure of main protease, followed by 3D pharmacophore filtration. Six top-ranked hits (ZINC58717986, ZINC60399606, ZINC58662884, ZINC45988635, ZINC54706757 and ZINC17320595) were identified based on their strong spatial affinity and forming H-bonds with key residues H41, E166, Q189 and T190 of the binding pocket of Mpro SARS-CoV-2. The 6 hits subjected to molecular dynamics simulations for 100 ns followed by binding free energy calculations using MM-PBSA technique. Interestingly, three hits showed free binding energy (ΔGbinding) lower than tert-butyl N-[1-[(2S)-1-[[(2S)-4-(benzylamino)-3,4-dioxo-1-[(3S)-2-oxopyrrolidin-3-yl]butan-2-yl]amino]-3-cyclopropyl-1-oxopropan-2-yl]-2-oxopyridin-3-yl]carbamate (α-ketoamide 13 b) (ΔGbinding) −76.67 ± 0.5 kJ/mol which suggested their potential against SARS-CoV-2. The best binding free energy candidates, ZINC58717986 (ΔGbinding) −98.41 ± 0.7 kJ/mol. The second-best hit candidate, ZINC54706757 (ΔGbinding) −83.4 ± 0.6 kJ/mol and the third one ZINC17320595 (ΔGbinding) −78.85 ± 0.5 kJ/mol. Per residue decomposition free energy indicates H41, S46, H164, E166, D187, Q189 and Q192 are hot spot residues while residues M49, M165, L167 and P168 contribute to the hydrophobic interactions. The pharmacokinetic study suggests that the selected 6 hits possess drug-like properties. The 3D pharmacophore virtual screening, molecular dynamics and MM-PBSA approaches facilitated identification 3 promising hits with low free binding energy as SARS-CoV-2 inhibitors.
Communicated by Ramaswamy H. Sarma
Graphical Abstract
Acknowledgements
This work used computational facilities of the Advanced Computing Research Centre, and Blue Crystal. University of Bristol, http://www.bris.ac.uk/acrc/. I thank the Advanced Computing Research Centre at the University of Bristol for access to High-performance Computing. The author is grateful to Dr. Richard Session at molecular modelling laboratory, University of Bristol for providing the computing resources and the access to BUDE program. Also, for his valuable scientific discussion with me about the virtual screening process.
Disclosure statement
The author declares no competing financial interest.