Figures & data
Table 1. Therapeutic agents regulating IDE expression and activity.
Figure 1. Possible side effects of pharmacological IDE inhibition. Pharmacological inhibition of IDE activity can increase levels of its targets – insulin, glucagon, amylin, and beta-amyloid (Aβ). Increased insulin acutely improves glucose tolerance; however, a long-term hyperinsulinemia can deteriorate insulin resistance. Increased activation of insulin-mediated proliferation pathway can facilitate cancerogenesis. Increased postprandial glucagon can induce gluconeogenesis in the liver which will worsen hyperglycemia. Increased levels of amylin in the pancreas and Aβ monomers in the brain can lead to the formation of toxic oligomers following by the function impairments and death of beta-cell and neurons.
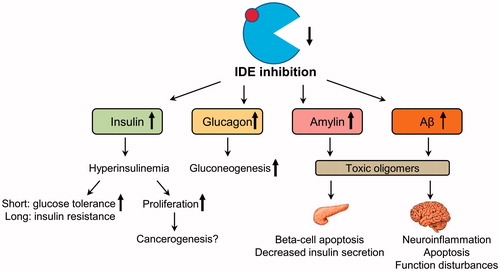
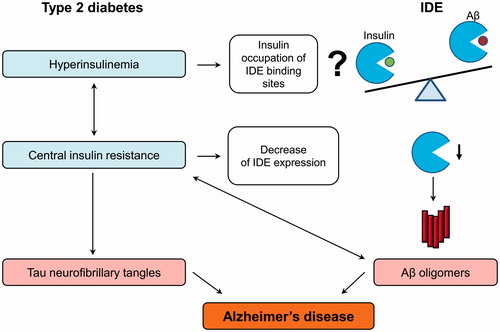
Figure 2. IDE as a pathological link between type 2 diabetes and Alzheimer’s disease. Type 2 diabetes is characterized by hyperinsulinemia and systemic insulin resistance. Insulin is hypothesized to decrease the IDE-mediated beta-amyloid (Aβ) degradation via the competitive inhibition, but this statement needs to be qualified. Insulin also increases Aβ production via other mechanisms, such as increased secretion. The central insulin resistance leads to the reduction of insulin signaling in the brain, which induces the hyperphosphorylation of tau protein and formation of toxic Aβ oligomers by the multiple mechanisms. Particularly, insulin resistance lowers the IDE expression and in this way decreases IDE-mediated Aβ degradation and additionally increases Aβ oligomer levels, which can in turn aggravate insulin resistance in the brain. All these molecular events finally lead to the formation of Aβ plaques and neurofibrillary tangles disturbing the neuronal organization and function.