Figures & data
Figure 1. Potential molecular pathways involved in inflammatory bowel disease(IBD) pathogenesis. In healthy individuals, bacterial invasions trigger cascades of immune events leading to chemokine secretion, bacterial clearance and apoptosis. Although there is much still to be discovered, genetic defects at three different levels are suspected to lead to chronic inflammation in IBD: 1) Mutations in environmental sensors and their signaling pathways, such as TLRs and CARD15, may impair the innate immune system and cause a decrease in NF‐kB activation and defensin production. 2) Mutations in transporters such as MDR1, OCTN1 and OCTN2, or in genes involved in epithelial integrity (DLG5, MYO9B) may affect the permeability of the epithelial barrier. 3) Mutations in genes involved in adaptive immunity, such as the human leukocyte antigen (HLA) genes and tumor necrosis factor (TNF‐) alpha within the major histocompatibility complex (MHC), as well as in genes coding for subunits of cytokines and cytokine receptors like IL‐12 and IL‐23R, may cause an imbalance between regulatory and effector cell immune responses important in the control of inflammatory reactions. IBD = inflammatory bowel disease; TLR = Toll‐like receptor; NF‐kB = nuclear factor‐kB; MDR1 = multidrug resistance gene; OCTN1 and 2 = organic cation transporter 1 and 2; DLG5 = drosophila discs large homolog 5.
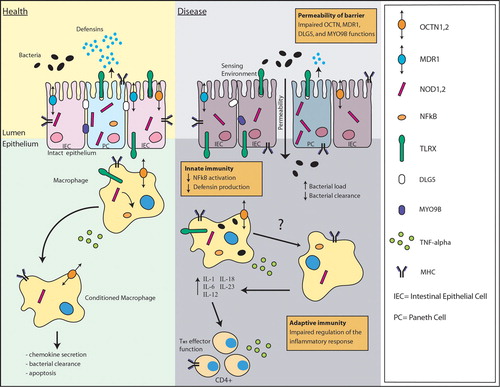
Figure 2. Schematic representation of the major steps towards the identification of genetic risk factors in disease. Once epidemiological studies have established a genetic link to disease, the search for genetic risk factors may progress through two major statistical approaches. Genome‐wide linkage analyses, which typically tests 300 simple sequence length polymorphism (SSLP) markers or 3000 single nucleotide polymorphisms (SNPs), can be used to identify chromosomal regions which putatively contain risk factors. Association analyses can be used to study these chromosomal regions or candidate genes identified through biological function. Recently, genome‐wide association analyses, which can test 100,000 to 500,000 SNPs, have also been used in the identification of risk loci. Association mapping narrows the search for risk factors to a particular genomic region, a specific gene or a single SNP. Once a putative association has been identified, and replicated in independent samples, the associated allele can then be tested for interaction with other identified genetic and non‐genetic risk factors. Re‐sequencing allows the identification of all variants in the region, in an attempt to identify the causal allele. The final, and often the most challenging, step in genetic studies is to link the genetic variant to a pathophysiological mechanism of underlying disease. The associated variant may be located within a specific gene and/or suggest a potential functional effect in disease etiology, but will also require functional studies in cellular and animal models, as well as evaluation in patient samples for final link to disease. The confirmation of novel disease risk factors should allow the identification of novel biological pathways to study in genetic association analysis, the generation of a molecular classification system for patients, and the discovery of potential drug targets for treatment.
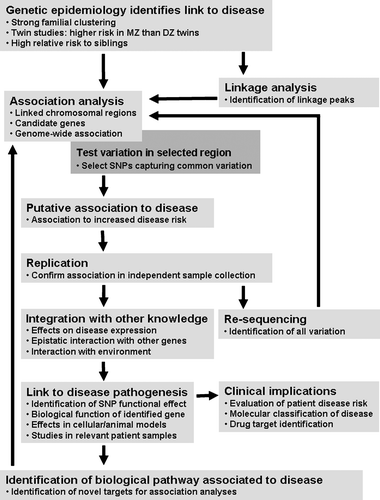
Figure 3. Classical representation of linkage and association studies. Linkage studies attempt to localize chromosomal regions containing disease genes by employing indirect statistical tests that examine for co‐inheritance of genetic markers and (disease) trait within families. Linkage analyses are very powerful to study highly penetrant genetic factors which are characteristically rare in a population. Typically, these studies look at many families and compare allele sharing between affected family members. For example, panel A depicts two parents and two affected children. Affected children are expected to share 0, 1, or 2 alleles in a proportion of, respectively, 0.25/0.50/0.25. If the affected siblings share alleles at a specific marker more often than they would by chance, then the marker is likely to be linked to the disease. Although the alleles shared between siblings have to be the same, the shared alleles across families can be different and still be linked to the disease. Association studies, on the other hand, test a specific allele across all individuals. There are two common designs for association studies: cases/controls (B), and trios consisting of two parents and one affected child (C). Case/control association studies test whether an allele is correlated with a disease by determining if allele frequencies differ between cohorts of unrelated cases and controls. Hundreds to thousands of samples are tested in cases/controls studies. Panel B illustrates an obvious difference between the frequencies of allele C in a study of six cases, 66%, and six controls, 33%, ‘suggesting’ association of this allele with the disease phenotype. In trios, the transmitted allele is equivalent in a case/control study to the ‘case’ and the untransmitted allele to the ‘control’. Panel C demonstrates the principle using a single trio, but hundreds of trios are usually tested. In this illustration, allele C is transmitted twice (100%) to the affected child ‘suggesting’ an association. Case/control studies are more efficient than family‐based designs because they require fewer samples (one case, one control as opposed to three samples in trios) to yield the same amount of information. However, in population‐based designs there are sometimes differences in allele frequencies in cases and controls not related to the trait under study. This phenomenon is called stratification. Trios are not prone to stratification since the perfectly matched control is the untransmitted chromosome. Linkage and association studies can be genome‐wide or targeted to a specific chromosomal region depending on the choice of markers. Fc = frequency of allele C; T = transmitted; U = untransmitted.
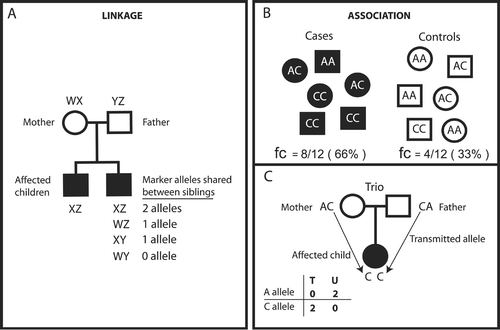
Table I. Evaluation of innate immunity in the genetic risk of inflammatory bowel diseases.