Figures & data
Figure 1. Endothelium‐dependent relaxation and endothelium‐dependent hyperpolarization. A: Left: The isometric tension developed by an arterial ring is studied in an organ chamber filled with thermostated and oxygenated physiological salt solution. The inset shows recording traces of an experiment reproducing the seminal observation first made by Furchgott and Zawadzki Citation4. An isolated ring of canine coronary artery with endothelium is contracted with prostaglandin F2α (PGF2α). The addition of acetylcholine produces a concentration‐dependent and endothelium‐dependent relaxation. Right: Bradykinin produces a concentration‐dependent and endothelium‐dependent relaxation in canine coronary arterial rings. The cyclooxygenase inhibitor, indomethacin, does not affect the relaxation to bradykinin while the NO synthase inhibitor L‐nitro‐arginine (L‐NA) produces a significant shift to the right of the concentration‐response curves, indicating a contribution of NO in the overall relaxation mechanism. The combination of the two inhibitors produces a further shift, indicating that prostacyclin contributes to the mechanism of bradykinin‐induced endothelium‐dependent relaxation when the NO synthase is inhibited. However, even in the presence of the two inhibitors, bradykinin still evokes the complete relaxation of the isolated arteries (modified from Citation210). SNP = sodium nitroprusside. B: Left: Changes in tension and in membrane potential are recorded in an isolated canine coronary artery with an intracellular microelectrode. Right: Acetylcholine produces a relaxation and a hyperpolarization of the vascular smooth muscle only when the endothelial cells are present (modified from Citation211).
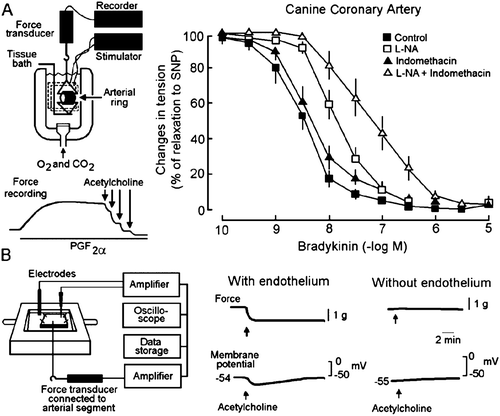
Figure 2. Endothelium‐dependent hyperpolarizations in the guinea‐pig carotid artery. A: Acetylcholine (ACh) induces an endothelium‐dependent hyperpolarization. B: In the presence of charybdotoxin (ChTX) plus apamin plus indomethacin plus L‐nitro‐arginine (L‐NA), acetylcholine no longer produces an endothelium‐dependent hyperpolarization but a small depolarization. Depol. = depolarization. C: In the presence of charybdotoxin (ChTX) plus apamin, acetylcholine produces an endothelium‐dependent hyperpolarization attributable to the release of NO and PGI2. PGI2 = prostacyclin. D: In the presence of charybdotoxin (ChTX) plus apamin plus indomethacin, acetylcholine produces an endothelium‐dependent hyperpolarization attributable to the release of NO. E: In the presence of charybdotoxin (ChTX) plus apamin plus L‐nitro‐arginine, acetylcholine produces an endothelium‐dependent hyperpolarization attributable to the release of PGI2. F: In the presence of indomethacin plus L‐nitro‐arginine (L‐NA), acetylcholine produces an EDHF‐mediated response. This response is not affected by the subsequent addition of a second NO synthase inhibitor, L‐monomethyl‐arginine (L‐NMMA) plus a NO scavenger, hemoglobin (middle trace), or another NO scavenger carboxy‐PTIO (lower trace), indicating that residual and stored NO are not involved.
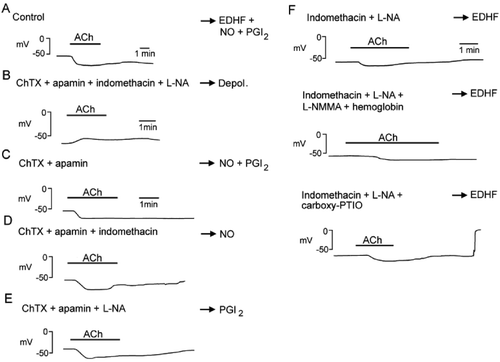
Figure 3. EDHF‐mediated responses. The activation of endothelial receptors (R) and the shear stress exerted by the flowing blood increase endothelial [Ca2+]i through calcium entry (TRP = transient receptor potential channel) and calcium release from internal stores (SR = sarcoplasmic reticulum). This activates endothelial KCa channels and produces endothelial hyperpolarization which is conducted through myoendothelial gap junctions to the underlying vascular smooth muscle. Cyclic‐AMP enhances the electrotonic conduction via gap junctions. Additionally, accumulation of potassium ions in the intercellular space can hyperpolarize the smooth muscle cells by activating KIR and Na+/K+‐ATPase. The presence of a ‘potassium cloud’ can markedly affect the contribution of K+ ions to EDHF‐mediated responses. Contractile agonists that stimulate their receptor on vascular smooth muscle cells increase intracellular calcium (Ca2+) concentration and depolarize the cells. The rise in intracellular calcium activates BKCa while the depolarization activates KV as a braking mechanism. Potassium effluxes through these two channels and accumulates in the intercellular space (potassium cloud). Potassium ions activate KIR and Na+/K+‐ATPase, preventing the effects of any subsequent rise of potassium during endothelium‐dependent hyperpolarization. The relative proportion of each mechanism depends on numerous parameters, including the state of activation of the vascular smooth muscle, the density of myoendothelial gap junctions, and the level of the expression of the appropriate isoforms of Na+/K+‐ATPase and/or KIR. BK = bradykinin; SP = substance P; SLIGRL‐NH2 = peptide agonist of proteinase‐activated recepor‐2; P3 = inositol trisphosphate; TRPV4 = transient receptor potential channel vanilloid 4; AC = adenylyl cyclase; αGA = glycyrrhetinic acid derivatives; CX = connexin; 4‐AP = 4‐aminopyridine; IbTX = iberiotoxin; ChTX = charybdotoxin; SK3 = small conductance calcium‐activated potassium channel formed by SK3 α subunits; IK1 = intermediate conductance calcium‐activated potassium channel formed by IK1 α subunits; Kir2.1 = Inward rectifying potassium channel constituted of Kir2.1 α subunits; KV = voltage‐gated potassium channels; BKCa = large conductance calcium‐activated potassium channels (modified from Citation58); ACh = acetylcholine.
![Figure 3. EDHF‐mediated responses. The activation of endothelial receptors (R) and the shear stress exerted by the flowing blood increase endothelial [Ca2+]i through calcium entry (TRP = transient receptor potential channel) and calcium release from internal stores (SR = sarcoplasmic reticulum). This activates endothelial KCa channels and produces endothelial hyperpolarization which is conducted through myoendothelial gap junctions to the underlying vascular smooth muscle. Cyclic‐AMP enhances the electrotonic conduction via gap junctions. Additionally, accumulation of potassium ions in the intercellular space can hyperpolarize the smooth muscle cells by activating KIR and Na+/K+‐ATPase. The presence of a ‘potassium cloud’ can markedly affect the contribution of K+ ions to EDHF‐mediated responses. Contractile agonists that stimulate their receptor on vascular smooth muscle cells increase intracellular calcium (Ca2+) concentration and depolarize the cells. The rise in intracellular calcium activates BKCa while the depolarization activates KV as a braking mechanism. Potassium effluxes through these two channels and accumulates in the intercellular space (potassium cloud). Potassium ions activate KIR and Na+/K+‐ATPase, preventing the effects of any subsequent rise of potassium during endothelium‐dependent hyperpolarization. The relative proportion of each mechanism depends on numerous parameters, including the state of activation of the vascular smooth muscle, the density of myoendothelial gap junctions, and the level of the expression of the appropriate isoforms of Na+/K+‐ATPase and/or KIR. BK = bradykinin; SP = substance P; SLIGRL‐NH2 = peptide agonist of proteinase‐activated recepor‐2; P3 = inositol trisphosphate; TRPV4 = transient receptor potential channel vanilloid 4; AC = adenylyl cyclase; αGA = glycyrrhetinic acid derivatives; CX = connexin; 4‐AP = 4‐aminopyridine; IbTX = iberiotoxin; ChTX = charybdotoxin; SK3 = small conductance calcium‐activated potassium channel formed by SK3 α subunits; IK1 = intermediate conductance calcium‐activated potassium channel formed by IK1 α subunits; Kir2.1 = Inward rectifying potassium channel constituted of Kir2.1 α subunits; KV = voltage‐gated potassium channels; BKCa = large conductance calcium‐activated potassium channels (modified from Citation58); ACh = acetylcholine.](/cms/asset/167fbc72-80ef-4740-8777-af2a5e8d2113/iann_a_248977_f0003_b.gif)
Figure 4. EDHF‐mediated responses in two strains of mice, wild‐type (C57BL/6), and NOS‐3 knockout mice (eNOS(−/−)). A: NOS‐3 knockout mice are hypertensive. B: Acetylcholine (ACh) produces an endothelium‐dependent relaxation in the isolated aortic rings of wild‐type mice (eNOS(+/+)) but no longer in that of NOS‐3 knockout mice (eNOS(−/−)). However, the NO donor and endothelium‐independent vasodilator sodium nitroprusside (SNP) produces a relaxation in the aorta from both strains. These results confirm the deletion of the NOS‐3 gene and demonstrate that, in a large blood vessel such as the aorta, NO is the preponderant endothelium‐derived relaxing factor released by acetylcholine. C: Acetylcholine induces a vasodilatation in the hindlimb of both wild‐type and NOS‐3 knockout mice. In the wild‐type mice, this vasodilatation is unaffected by a depolarizing solution (KCl) and partially inhibited by the combination of charybdotoxin (CTX) and apamin (apa) but not by charybdotoxin alone, indicating that an EDHF‐mediated response may contribute to the mechanism of vasodilatation but is not the preponderant mechanism. In the NOS‐3 knockout mice, the acetylcholine‐induced vasodilatation is preserved and is blocked by KCl or the combination of charybdotoxin plus apamin, partially inhibited by charybdotoxin alone and unaffected by the combination of iberiotoxin (IbTX), apamin, glibenclamide (Glib) and 4‐aminopyridine (4AP). These results indicate that acetylcholine induces an endothelium‐dependent vasodilatation showing the hallmarks of EDHF‐mediated responses. The EDHF‐mediated response compensates the disruption of the NOS‐3 gene. D: Original recording of arterial blood pressure in an anaesthetized NOS‐3 knockout mouse treated with an inhibitor of cyclooxygenase, diclofenac, and an inhibitor of NO synthase, L‐arginine‐methyl‐ester (L‐NAME). The intravenous administration of the endothelium‐dependent vasodilator bradykinin induces a transient decrease in arterial blood pressure. The summary bar graphs show the concentration‐dependent changes in mean arterial pressure (MAP) evoked by administration of bradykinin in wild‐type and eNOS(−/−) mice under control conditions (top graphs) or after treatment with inhibitors of cyclooxygenases and NO synthases (lower graphs). The amplitude of the hypotensive response induced by bradykinin is similar in NOS‐3 knockout and wild‐type animals. In both strains, the effect of bradykinin is unaffected by the presence of inhibitors of cyclooxygenases and NO synthases. These data strongly suggest that EDHF‐mediated responses play an essential role in the hypotensive effect of bradykinin (modified from Citation159). CTL = control.
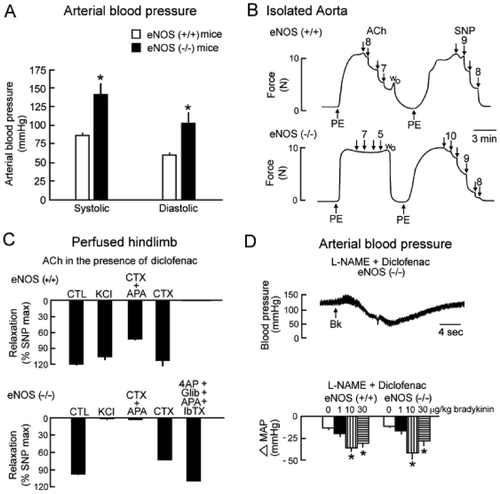
Figure 5. Endothelium‐dependent hyperpolarizations in isolated human coronary arteries (in the presence of inhibitors of cyclooxygenases and NO synthases). A: In coronary arteries taken from a young patient (15 months old, congenital heart disease), bradykinin produces an endothelium‐dependent hyperpolarization. B: In coronary arteries taken from an older patient (64 years old, ischemic heart disease) the endothelium‐dependent hyperpolarization to bradykinin is reduced markedly when compared to that observed in the juvenile patient. C: In coronary arteries taken from a patient (68 years old, ischemic cardiomyopathy) the endothelium‐dependent hyperpolarization to bradykinin is virtually absent. The presence of the angiotensin‐converting enzyme inhibitor, perindoprilat, restores the endothelium‐dependent hyperpolarization (modified from Citation1).
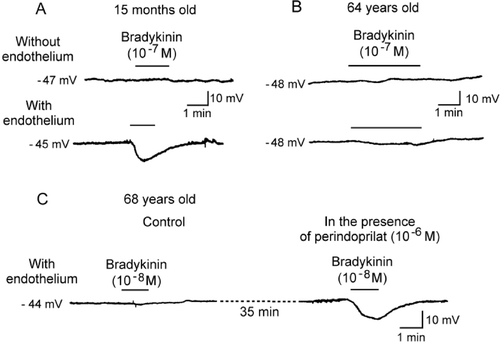
Table I. Nonexhaustive summary of potential mechanisms underlying endothelium‐dependent hyperpolarizations/relaxations resistant to the combined presence of inhibitors of NO‐synthases and cyclooxygenases.