Figures & data
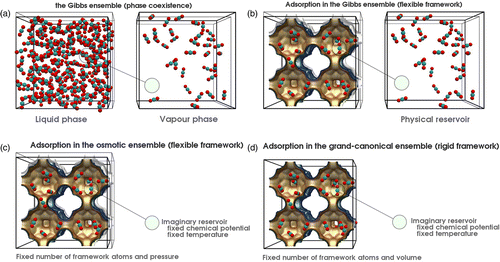
![Figure 1 (Colour online) Ensembles: Shown are the eight ensembles for a single component system. The systems interact through a combined temperature, pressure and chemical potential reservoir. The ensembles on the left are adiabatically insulated from the reservoir, whereas those on the right are in thermal contact with the reservoir. Pistons and porous walls allow for volume and particle exchange. Adiabatic walls are shown cross-hatched, whereas dithermal walls are shown as solid lines. Ensembles on the same height are related by Laplace and inverse Laplace transformations. The pressure stands for the pressure and the tension. Picture taken from Ref. [Citation108].](/cms/asset/d1e102e0-862b-4389-93e6-b6dbc4038086/gmos_a_819102_f0001_oc.gif)
![Figure 6 (Colour online) NS, (left) first-order transitions are dealt with differently by parallel tempering, Wang–Landau sampling, and NS (right). NS is a top-down approach where a set of random walkers converge downwards in energy at a rate equal to the logarithm of the phase space. Left figure taken from Ref. [Citation256].](/cms/asset/83fcc237-08e5-4cea-afee-b455dda71e0e/gmos_a_819102_f0006_oc.gif)
Table 1 Energies of a snapshot of 20 CO2 and 10 pentane molecules (TraPPE model) in a  Å box computed with RASPA, TINKER and DLPOLY 4.
Å box computed with RASPA, TINKER and DLPOLY 4.
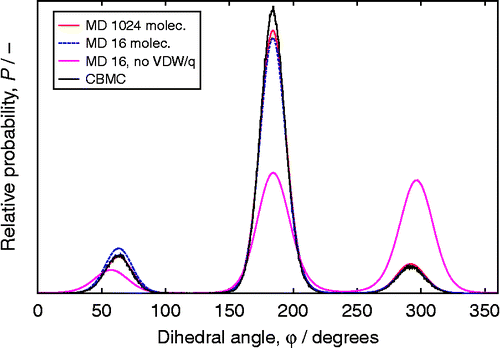
Table 2 Comparison of CBMC and CFMC for adsorption of CO2 and CH4 in MFI, as a function of fugacity f.
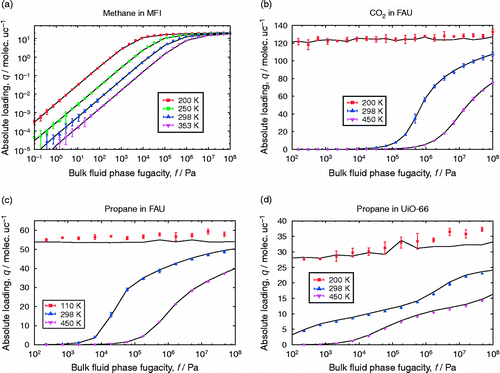
Table 3 Gibbs simulations of TraPPE ethane.
Table 4 Gibbs simulations of TraPPE CO2.
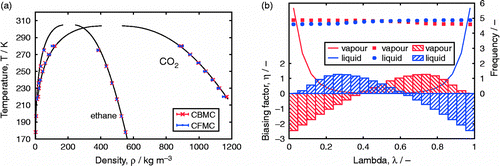
Table 5 CBMC versus CFMC for an equimolar mixture of CO2/N2 in DMOF at 300 K, with and without the identity switch move.
Supplemental material