
Figures & data
Figure 1. (Colour online) Schematic representation of: (a) test molecule insertions, often resulting in overlaps in dense phases due to the lack of cavities large enough to accommodate test molecules. (b–d) Gradual insertion or removal of a fractional molecule by performing random walks in λ-space. At , the fractional molecule does not interact with the other ‘whole’ molecules (ideal gas behaviour) and at
, the fractional molecule interacts fully with other whole molecules. By performing thermalisation trial moves e.g. translations, rotations, volume moves, etc., the other whole molecules can adapt to the value of λ.
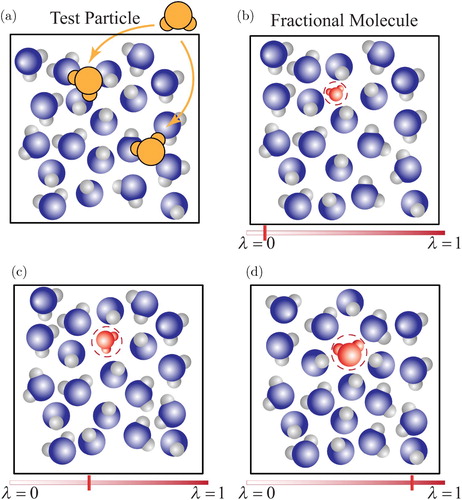
Table 1. A historical overview of the most important methods and developments leading to advancements in staged insertions and free energy calculations in the framework of expanded ensembles.
Figure 2. (Colour online) MC trial moves in the CFCMC Gibbs ensemble are used to perform stepwise molecule exchanges between the phases [Citation123]. The coupling parameter λ is used to scale the interactions of the fractional molecule with surrounding molecules. The deletion or insertion of the fractional molecule is staged using the coupling parameter λ. Trial moves to facilitate molecule exchanges are: (a) Random walks in λ-space: attempts to randomly change the value of λ while keeping the positions of all molecules constant. For the cases or
, the trial move is rejected. (b) Swapping the fractional molecule: the fractional molecule is removed from one simulation box and inserted at a randomly selected position in the other simulation box. The value of λ is not changed in this trial move. (c) Identity changes: an attempt to transform the fractional molecule into a whole molecule, accompanied by changing a randomly selected molecule in the other simulation box into a fractional molecule, while keeping the positions of all molecules fixed. For these trial moves, the acceptance rules are derived in Ref. [Citation123].
![Figure 2. (Colour online) MC trial moves in the CFCMC Gibbs ensemble are used to perform stepwise molecule exchanges between the phases [Citation123]. The coupling parameter λ is used to scale the interactions of the fractional molecule with surrounding molecules. The deletion or insertion of the fractional molecule is staged using the coupling parameter λ. Trial moves to facilitate molecule exchanges are: (a) Random walks in λ-space: attempts to randomly change the value of λ while keeping the positions of all molecules constant. For the cases λ<0 or λ>1, the trial move is rejected. (b) Swapping the fractional molecule: the fractional molecule is removed from one simulation box and inserted at a randomly selected position in the other simulation box. The value of λ is not changed in this trial move. (c) Identity changes: an attempt to transform the fractional molecule into a whole molecule, accompanied by changing a randomly selected molecule in the other simulation box into a fractional molecule, while keeping the positions of all molecules fixed. For these trial moves, the acceptance rules are derived in Ref. [Citation123].](/cms/asset/c1faf15b-3d7b-4f09-916c-fd4585a16274/gmos_a_1828585_f0002_oc.jpg)
Figure 3. (Colour online) Chemical potentials of water (TIP3P [Citation179]) and hydrogen (Marx [Citation180]) from vapour-liquid equilibrium simulations of binary water-hydrogen mixtures in the CFCMC Gibbs ensemble, as a function of pressure, at T=323 K (squares), T=366 K (diamonds), T=423 K (triangles). (a) Chemical potential of water in the liquid phase (closed symbols) and chemical potential of water in the gas phase (open symbols). (b) Chemical potential of hydrogen in the liquid phase (closed symbols) and chemical potential of hydrogen in the gas phase (open symbols). Lines and dashed lines are used as a guide for the eye for the liquid phase and gas phase, respectively. The chemical potentials in this figure are part of the previously published VLE data of water-hydrogen of Ref. [Citation178]. These chemical potentials were not explicitly reported in Ref. [Citation178]. Simulation details on the phase coexistence of the water-hydrogen system are provided in Ref. [Citation178]. Raw data and uncertainties are provided in the Supporting Information. (c) Molecule exchanges between the liquid and the gas phase are performed in a gradual manner using the fractional molecules of water and hydrogen (marked molecules). The snapshot is generated using iRASPA [Citation185].
![Figure 3. (Colour online) Chemical potentials of water (TIP3P [Citation179]) and hydrogen (Marx [Citation180]) from vapour-liquid equilibrium simulations of binary water-hydrogen mixtures in the CFCMC Gibbs ensemble, as a function of pressure, at T=323 K (squares), T=366 K (diamonds), T=423 K (triangles). (a) Chemical potential of water in the liquid phase (closed symbols) and chemical potential of water in the gas phase (open symbols). (b) Chemical potential of hydrogen in the liquid phase (closed symbols) and chemical potential of hydrogen in the gas phase (open symbols). Lines and dashed lines are used as a guide for the eye for the liquid phase and gas phase, respectively. The chemical potentials in this figure are part of the previously published VLE data of water-hydrogen of Ref. [Citation178]. These chemical potentials were not explicitly reported in Ref. [Citation178]. Simulation details on the phase coexistence of the water-hydrogen system are provided in Ref. [Citation178]. Raw data and uncertainties are provided in the Supporting Information. (c) Molecule exchanges between the liquid and the gas phase are performed in a gradual manner using the fractional molecules of water and hydrogen (marked molecules). The snapshot is generated using iRASPA [Citation185].](/cms/asset/b1662d98-f4d0-466e-80be-fc6d53432d88/gmos_a_1828585_f0003_oc.jpg)
Figure 4. (Colour online) Trial moves associated with fractional molecules facilitating chemical reactions in the reaction ensemble. The ammonia synthesis reaction is considered as an example to demonstrate the trial moves: (a) Random walks in λ-space: an attempt to change the value of λ while orientations and positions of all molecules are fixed. Here, the coupling parameter is used to scale the interactions of
. (b) Reaction for the fractional molecules; the fractional molecules of reaction products
are inserted and the fractional molecules of the reactants
are removed. The orientations and positions of other molecules are unchanged. (c) Reaction for the whole molecules; randomly selected reaction products (
) are transformed into fractional molecules and the fractional molecules of the reactants
are transformed into whole molecules. The acceptance rules for these trail moves are provided in Ref. [Citation61].
![Figure 4. (Colour online) Trial moves associated with fractional molecules facilitating chemical reactions in the reaction ensemble. The ammonia synthesis reaction N2+3H2⇌2NH3 is considered as an example to demonstrate the trial moves: (a) Random walks in λ-space: an attempt to change the value of λ while orientations and positions of all molecules are fixed. Here, the coupling parameter is used to scale the interactions of N2+3H2. (b) Reaction for the fractional molecules; the fractional molecules of reaction products 2NH3 are inserted and the fractional molecules of the reactants N2+3H2 are removed. The orientations and positions of other molecules are unchanged. (c) Reaction for the whole molecules; randomly selected reaction products (2NH3) are transformed into fractional molecules and the fractional molecules of the reactants N2+3H2 are transformed into whole molecules. The acceptance rules for these trail moves are provided in Ref. [Citation61].](/cms/asset/469f6ae3-8212-4a80-8e2f-47e9be7ec1cf/gmos_a_1828585_f0004_oc.jpg)
Figure 5. (Colour online) (a) Mole fractions of ammonia at chemical equilibrium obtained from Rx/CFC simulations (symbols) and experiments (solid lines) [Citation188] at 573 K (circles), 673 K (upward-pointing triangles), 773 K (squares) and, 873 K (downward-pointing triangles) as a function of pressure. All simulations were started from a random configuration of 120 and 360
molecules and no ammonia molecules. (b) Fugacity coefficients of ammonia at equilibrium obtained from Rx/CFC simulations (symbols) using Equation (Equation13
(13)
(13) ) and the Peng-Robinson EoS (lines) [Citation24] at 573 K (circles), 673 K (upward-pointing triangles), 773 K (squares) and, 873 K (downward-pointing triangles) as a function of pressure. Raw data is taken from Ref. [Citation61].
![Figure 5. (Colour online) (a) Mole fractions of ammonia at chemical equilibrium obtained from Rx/CFC simulations (symbols) and experiments (solid lines) [Citation188] at 573 K (circles), 673 K (upward-pointing triangles), 773 K (squares) and, 873 K (downward-pointing triangles) as a function of pressure. All simulations were started from a random configuration of 120 N2 and 360 H2 molecules and no ammonia molecules. (b) Fugacity coefficients of ammonia at equilibrium obtained from Rx/CFC simulations (symbols) using Equation (Equation13(13) ∏i=1Rϕi−νi=p(λR=1)p(λR=0)×Zmξ(13) ) and the Peng-Robinson EoS (lines) [Citation24] at 573 K (circles), 673 K (upward-pointing triangles), 773 K (squares) and, 873 K (downward-pointing triangles) as a function of pressure. Raw data is taken from Ref. [Citation61].](/cms/asset/0586dfee-4781-4fbb-918b-5112cea20e29/gmos_a_1828585_f0005_oc.jpg)
Figure 6. (Colour online) Reaction enthalpy of the Haber-Bosch process per mole of at pressures between 100 bar and 800 bar, at T=573 K. The reaction enthalpy at P=1 bar is indicated with an arrow on the left hand side of the figure. The contribution of intermolecular interactions to the reaction enthalpy is computed using: Peng-Robinson EoS (upward-pointing triangles); PC-SAFT (downward-poiting triangles); CFCNPT ensemble (circles). No binary interaction parameters were used in the EoS modelling. The data is taken from Ref. [Citation125].
![Figure 6. (Colour online) Reaction enthalpy of the Haber-Bosch process per mole of N2 at pressures between 100 bar and 800 bar, at T=573 K. The reaction enthalpy at P=1 bar is indicated with an arrow on the left hand side of the figure. The contribution of intermolecular interactions to the reaction enthalpy is computed using: Peng-Robinson EoS (upward-pointing triangles); PC-SAFT (downward-poiting triangles); CFCNPT ensemble (circles). No binary interaction parameters were used in the EoS modelling. The data is taken from Ref. [Citation125].](/cms/asset/d0050be6-f430-4136-a6cb-6dab3a31bdae/gmos_a_1828585_f0006_oc.jpg)
Figure 7. (Colour online) Probability distribution for pure methanol obtained from CFCNPT simulation at T=298 K and P=1 bar (410 molecules). Equation (Equation17
(17)
(17) ) is used to compute
for temperatures between 329 and 273 K from a single simulation at 298 K. The bold red line indicates the Boltzmann distribution of
. The data is taken from Ref. [Citation127].
![Figure 7. (Colour online) Probability distribution p(λ,T) for pure methanol obtained from CFCNPT simulation at T=298 K and P=1 bar (410 molecules). Equation (Equation17(17) pλ,P=c⋅δ(λ′−λ)expβVP⋆−PP⋆(17) ) is used to compute p(λ,T) for temperatures between 329 and 273 K from a single simulation at 298 K. The bold red line indicates the Boltzmann distribution of p(λ,T=298K). The data is taken from Ref. [Citation127].](/cms/asset/04959c4c-793e-4689-b6b3-bfebb7432f5d/gmos_a_1828585_f0007_oc.jpg)