Figures & data
Table 1. Isolation information for new Nitzschia strains sequenced for this study. Original strain designations correspond to the isolate names on the voucher slides held in Royal Botanic Garden of Edinburgh. See for explanation of ‘N. palea agg.’.
Table 2. GenBank accession numbers for the species and strains analysed. Accession numbers for sequences obtained for this study are shown in bold. Strain identifier refers to the names of the isolates as used in this paper. Within the Nitzschia palea species complex, clones are identified according to their membership of one of the three putative species (OTUs) recognized by Trobajo et al. (Citation2009), i.e. ‘LSU clade A’, ‘mating group 1’ and ‘mating group 2’, or, for clones not belonging to any of these groups, they are referred to as ‘N. palea agg.’.
Table 3. p distances (right-top) and bp differences (left-bottom) between N. palea cox1 sequences (alignment of 495 bp). Clades with good support or identical sequences are outlined by a solid line. Shaded boxes: p distance and bp difference for the maximally different N. palea clones (UK vs New Scot2).
Fig. 1. ML tree based on cox1 gene, illustrating the phylogeny of N. palea sequences. The tree was rooted using two Sellaphora pupula sequences. Maximum parsimony, neighbour joining and maximum likelihood bootstrap values above 50% and BI posterior probabilities above 95% (MP/ NJ/ ML/ BI) are shown on the branches. Internal nodes supported by bootstrap values above 80% and posterior probabilities above 95% in at least two types of analyses are highlighted as thick lines.
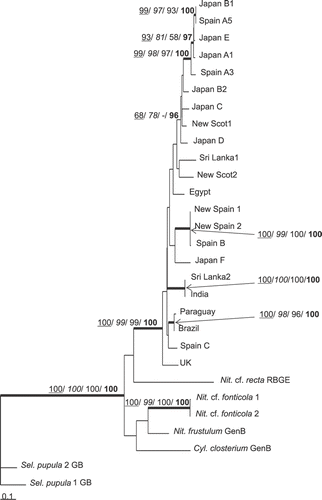
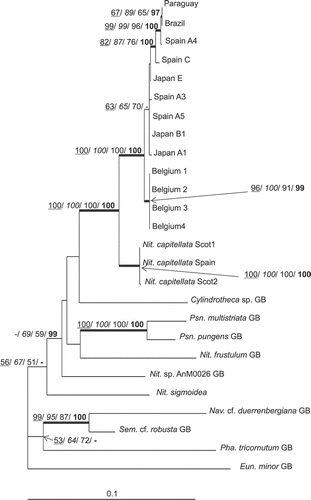
Fig. 2. ML tree based on rbcL gene, illustrating the phylogeny of N. palea sequences. The tree was rooted using Eunotia minor. Maximum parsimony, neighbour joining and maximum likelihood bootstrap values above 50% and BI posterior probabilities above 95% (MP/ NJ/ ML/ BI) are shown on the branches. Internal nodes supported by bootstrap values above 80% and posterior probabilities above 95% in at least two types of analyses are highlighted as thick lines.