Figures & data
Table 1. Overview of different inherited thrombocytopenias.
Table 2. Overview of inherited platelet function disorders associated with a normal platelet count. The conditions listed have a normal platelet size.
Table 3. Likelihood of experiencing bleeding for commonly encountered platelet function disorders.
Table 5. International Society on Thrombosis and Haemostasis recommended investigations for suspected inherited platelet function disorders.
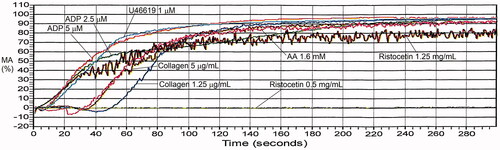
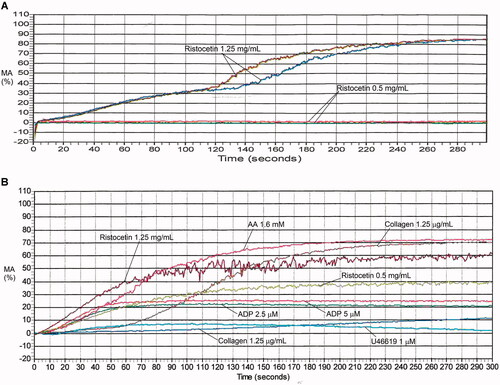
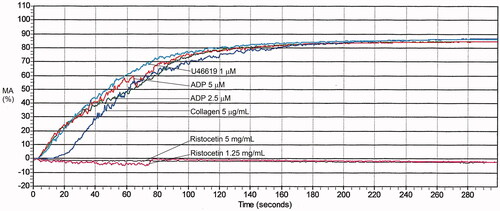
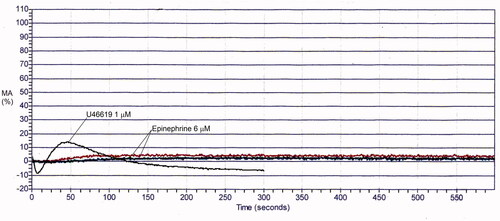
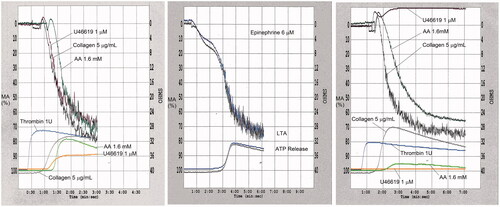
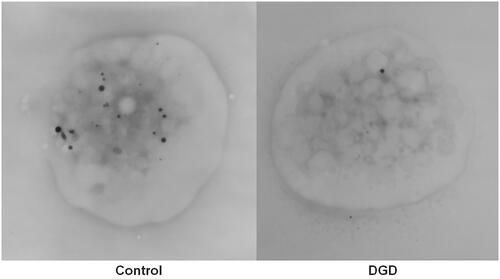
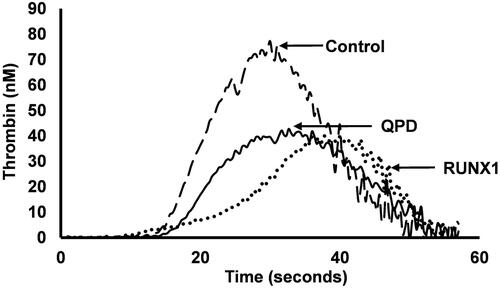
Table 6. Maximal aggregation findings for representative cases with platelet function disorders, evaluated by light transmission platelet aggregometry.
Table 7. International recommendations on preanalytical variables for platelet aggregation studies by light transmission aggregometry.
Table 8. International recommendations on analytical variables for platelet aggregation studies by light transmission aggregometry.
Table 9. Summary of recommended agonists for platelet aggregation studies by different international guidelines.
Table 10. Approach to interpreting platelet aggregation studies by light transmission aggregometry. The approach has been updated from published guidance [Citation8].
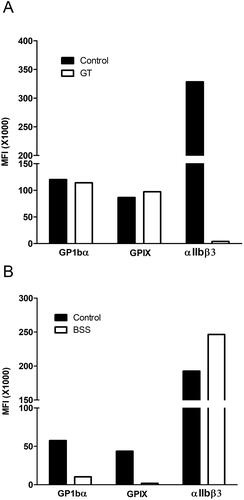
Table 11. Summary of key consensus recommendations from the International Society on Thrombosis and Haemostasis on using flow cytometry for the assessment of disorders of platelet number and function [Citation311].

![Figure 1. Causes of platelet function disorders and inherited thrombocytopenias. The figure summarizes the genes and pathways that contribute to the pathogenesis of platelet disorders, which includes regulatory genes that have impacts on the early or later stages of megakaryopoiesis, as summarized in Lentaigne et al. [Citation13].](/cms/asset/44eda53c-64e4-47b8-baeb-a607297076de/ilab_a_2049199_f0001_c.jpg)