
Figures & data
Table 1. Studies associated with periodontal pathogens and GIT dysfunction.
Figure 1. Schematic representation explaining the link between periodontal disease, oral bacteria, and gastrointestinal (GIT) dysfunction: (A) the oral cavity is a reservoir of millions of microbes that form a biofilm on the hard (teeth and restorations) and soft tissues (gums, tongues, palate, etc). The interaction of the microbes to the periodontal tissues initiates an inflammatory response that locally leads to periodontal inflammation; (B) the onset of periodontal inflammation (gingivitis and periodontitis) is marked by a massive release of proinflammatory mediators such as IL1, IL8, IL17, and TNF along with activation of various signaling pathways and host receptors such as toll-like receptors (TLRs), the complement system, nuclear factor kappa beta (NF-Kb); (C) these proinflammatory mediators enter the systemic circulation via blood vessels or via the saliva and reach the liver where they trigger the acute phase response and the release of acute phase proteins like C-reactive proteins (CRP). This increases the systemic inflammatory burden and oxidative stress. (D) The increased inflammatory burden in turn affects various parts of the gut and leads to GIT dysfunction (Created in Biorender).
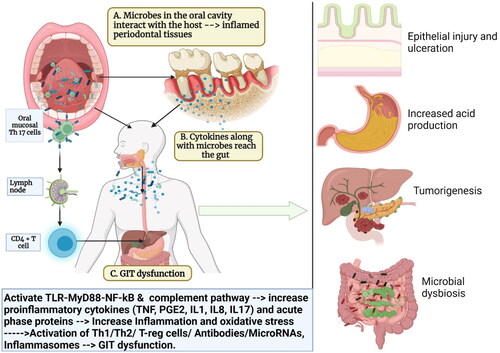
Figure 2. Schematic representation of how oral bacteria and their by-product injure the epithelium, increase gut permeability, and increase the risk of gut dysbiosis and inflammation: Oral bacteria interact with toll-like receptors (TLRs) in the gut and causes the release of proinflammatory cytokines (interleukins (IL1, IL8), prostaglandins (PGE2), tumor necrosis factors (TNF). Some of the oral bacteria also release volatile sulfur such as hydrogen sulfide, dimethyl sulfide, dimethyl disulfide, and methyl mercaptan which causes a reversible increase in epithelial permeability and loss of barrier function. Hydrogen sulfide has also been shown to increase crypt formation and ulceration by inducing DNA hypomethylation in the gut mucosa. These inflammatory mediators also affect the cell adhesion molecules such as zonula occludens and occludins and increase the permeability. This facilitates the easy invasion of the bacteria further into the gut tissues, increasing the risk of microbial dysbiosis in gut and gut inflammation (Created in Biorender).
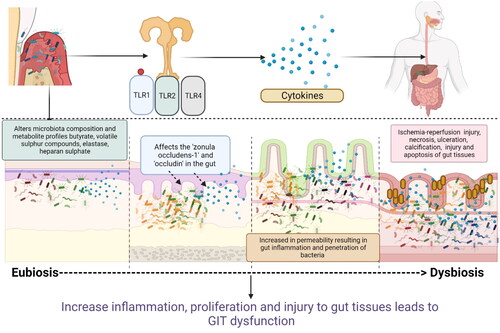
Figure 3. Schematic representation of how periodontal pathogens activate various immune cells and increase gut inflammation: Periodontal pathogens activate the various signaling pathways of inflammations such as nuclear factor kappa beta (NF-kB), which in turn activates the expression of co-stimulatory surface molecules (CD80 and CD86) which in turn activates various subsets of T cells (T-helper cells, T-suppressor cells, T-regulatory cells) and antigen-presenting cells (macrophages, dendritic cells, and langerhans cells) in the host. Periodontitis activation of the complement system with the release of C3a and C5a (chemoattractant). Increased release of chemoattractant (IL8, TNF, C3a, C5a) induces the influx of neutrophils and macrophages into the gut tissues. These mediators also trigger Th1 responses (induce the release of IL-12, IL1, IFN, TNF alpha, and IL-18) and Th2 response (IL-4, IL-5, IL-13 and IL-13). These cytokines along with antibodies produce mucosal inflammation, increase gastric permeability, change gastric motility, delay gastric emptying, and produce symptoms of colitis in patients with IBD, CD, and UC. T-helper cells lead to the formation of plasma cells that produce antibodies (IgG, IgA, and IgM antibodies). Increased levels of IgA and IgG antibodies have been linked with gut inflammation and the development of GIT disorders such as IBD (Created in Biorender).
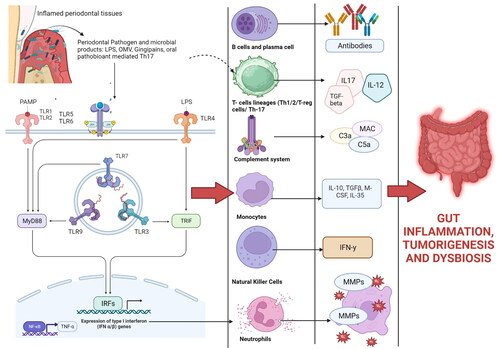
Figure 4. Periodontal tissues are massive producers of Th-17 cells which undergo pathogenic conversion in the gut, and this causes IFN-γ-producing Th1-like CD4+ T cells. Some oral bacteria that enter the gut increases the conversion of oral Th17 TEM cells into the IFN-γ-producing Th17/Th1 cell (pathogenic Th17 cells). Gut-migrated oral pathogenic Th17 cells in turn increase the inflammation in the gut and activate the gut-colonized oral microbiota, exacerbating colitis and other GIT diseases. Evidence has shown that oral pathobiont-reactive T cells that arise during periodontitis are colitogenic and are a potential cause of many GIT disorders such as inflammatory bowel disease (IBD) (Created in Biorender).
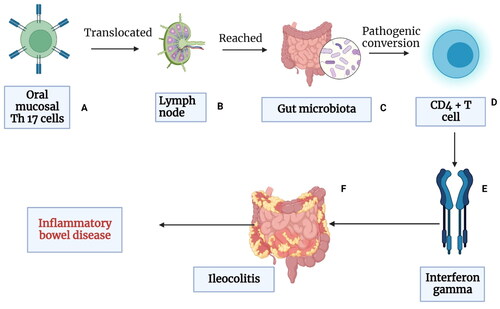
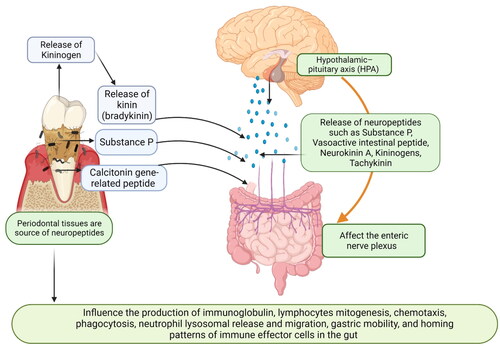
Figure 5. Schematic representation explaining how periodontal tissues are linked to the Central nervous system and gut: the Central nervous system (CNS) is linked with the gut via the hypothalamic–pituitary axis (HPA). Various neuropeptides such as substance P, vasoactive intestinal peptide (VIP), calcitonin gene-related peptides, neurokinin A (NKA), kininogens, and tachykinin serve as a major link between the enteric nervous system (ENS), CNS, and gut. Periodontal bacteria also increase the release of kininogens and substance P. These neuropeptides affect the enteric nervous systems (vagal and mesenteric nerves and affect gut mobility, gastric acid secretion, and release of inflammatory mediators. These neuropeptides act as intracellular signaling molecules and directly influence the production of immunoglobulin, lymphocyte mitogenesis, chemotaxis, phagocytosis, neutrophil lysosomal release and migration, and homing patterns of immune effector cells in the gut (created in Biorender).
Supplemental Material
Download MS Word (42.9 KB)Data availability statement
Data is available upon request via email to the corresponding author.