

GRAPHICAL ABSTRACT
ABSTRACT
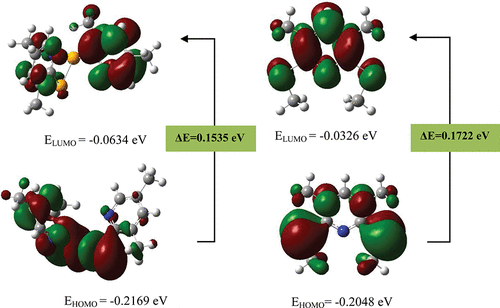
The present study explores various quantum chemical models to better understand the molecular properties of pyridylselenium compounds. We have carried out theoretical and experimental analyses of bis(3,5-dimethyl-2-pyridyl)diselenide (1), 3,5-dimethyl-2,6-bis(methylselenenyl) pyridine (2) and 3,5-dimethyl-2-(methylselenenyl)pyridine (3). The study demonstrates the effect of structural variations on molecular properties of these compounds. Density functional theory (DFT) has been used to calculate vibrational wavenumbers and intensity of vibrational bands and the results obtained are in excellent agreement with the experimental FT-IR data. The 1H and 13C NMR chemical shifts were calculated using GIAO method in gas phase and solvent phase of CDCl3 by taking IEFPCM method and correlations have been developed which accurately predicts the chemical shift values of various organoselenium compounds. The HOMO-LUMO energies in the gas and solvent phase were calculated by using B3LYP/6-311++G(d,p) level of theory. The HOMO-LUMO energy gaps in different molecules were compared and correlated with their biological activity.
Acknowledgments
The authors thank Dr. P.V. Bharatam, NIPER, Mohali and M. Govindarajan, Department of Physics, MGGA College, Mahe, India for providing useful insight during the course of this work.