
Abstract
Structural and anion binding studies have provided an insight into the conformational role of an allosteric CuII ion in the trinuclear catalyst (L 2–2H) {\rm Cu_{3}^{II}} . L 2 is a novel trinucleating ligand with pyridyl, pyrimidyl and amide donor groups. (L 2–2H)Cu (1) has been characterized by X-ray crystallography. The Cu ion is located at the allosteric site and coordinated by two amide N and two pyrimidine N atoms, the CuN4 plane is tetrahedrally distorted, and the complex adopts a helically twisted conformation. In contrast, the (L 2–2H)Cu3(μ4-C2O4) subunit of the dodecanuclear complex [(L 2–2H)4Cu12(μ4-C2O4)2(μ-OH)4(μ-Cl)4Cl4(H2O)2] (5) is roof-shaped, and the allosteric Cu is located on the top of a square-based pyramid. The oxalate coligand is coordinated by the two catalytic Cu ions in an unusual 1,4-μ-O,O bridging mode with an O ⃛O “bite length” of 2.6 Å and a Cu ⃛Cu distance of 6.4 Å. Intramolecular transesterification of the phosphodiester 2-(hydroxypropyl)-p-nitrophenyl phosphate (HPNP) by [(L 2–2H)Cu3]4+ was investigated, in comparison with the closely related complex [(L 3–2H)Cu3]4+ in which the ligand framework is somewhat less flexible. From a kinetic analysis of cleavage rate at varying HPNP concentrations, K HPNP (the equilibrium constant for binding of HPNP to the complex) and k cat (first-order rate constant for cleavage of HPNP when bound to the complex) parameters were derived: K HPNP=190 M-1 ([(L 2–2H)Cu3]4+) and 305 M-1 ([(L 3–2H)Cu3]4+), k cat=10×10-3 s-1 ([(L 2–2H)Cu3]4+) and 3.3×10-3 s-1 ([(L 3–2H)Cu3]4+). Anion binding constants of the complexes were determined by monitoring competitive inhibition of HPNP cleavage. The complexes have a high affinity to {\rm SO_{4}^{2-}} , {\rm HPO_{4}^{2-}} , and {\rm ReO_{4}^{-}} , which appear to be of the appropriate size for bridging coordination, while “smaller” anions {\rm CH_{3}CO_{2}^{-}} and {\rm HCO_{3}^{-}} are bound less efficiently. [(L 3–2H)Cu3]4+ has a higher affinity than [(L 2–2H)Cu3]4+ to HPNP but a lower affinity to the rather large anion {\rm ReO_{4}^{-}} . This is interpreted as a consequence of the reduced flexibility of [(L 3–2H)Cu3]4+, which slightly disfavours widening of the Cu ⃛Cu distance for incorporation of perrhenate. Similarly, the somewhat lower reactivity of [(L 3–2H)Cu3]4+ is attributed to the larger energy gap between the ground state and sterically more demanding (and less efficiently stabilized) transition state.
Abstract
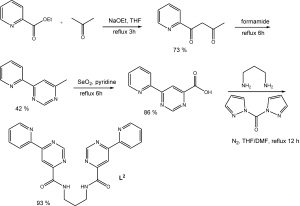
Synthesis of L 2.
Intramolecular cleavage of the phosphodiester 2-(hydroxypropyl)-p-nitrophenyl phosphate (HPNP).
Proposed mechanism of HPNP cleavage by [(L n –2H)Cu3]4+, including estimated O ⃛O distances of bridging HPNP in the ground and transition state.
![Proposed mechanism of HPNP cleavage by [(L n –2H)Cu3]4+, including estimated O ⃛O distances of bridging HPNP in the ground and transition state.](/cms/asset/1224691e-6594-4689-bf77-789e7e3ccf4b/gsch_a_31030_o_f16.gif)
Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft (Gerhard Hess-Programm) and supported by the Fonds der Chemischen Industrie.
Notes
†Crystallographic data (excluding structure factors) for the structure included in this paper have been deposited with the Cambridge Crystallographic Crystallographic Data Centre as supplementary publication no. CCDC-129330 (L 3), CCDC-192329 (5), CCDC-192328 (1). Copies of the data can be obtained free of charge on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK. Fax: +44-1223-336-033; E-mail: [email protected].
The Future of Supramolecular ChemistrySelf-assembly involving coordination chemistry has recently undergone a particularly tremendous development, making available many fascinating complex structures by relatively simple and rapid synthetic procedures. Much has been learned about the use of directional bonding afforded by metal centres for the rational assembly of supramolecular architectures. In view of recent breakthroughs in the engineering of prototypical molecular scale electronic circuits, it is a very exciting challenge to create nanosized structures by self-assembly and to individually control their physical properties for “bottom up” solutions of problems to miniaturisation.Roland Krämer (born 1963) undertook undergraduate studies at the universities of Karlsruhe and Munich. He received his PhD in Munich 1991 in Professor W. Beck's laboratory (organometallic chemistry) and was a postdoc 1991–1992 in the lab of J.-M. Lehn at Strasbourg (inorganic self-assembly). In 1997, he finished his Habilitation at University of Münster (bioinorganic Chemistry) and became a professor at the University of Heidelberg in 1999. His research interests include coordination, bioinorganic and bioconjugate chemistry.
