Figures & data
Figure 1. Aberrant splicing alterations in ALS. Loss of nuclear RNA binding proteins (RBP), including splicing factors, may lead to perturbed pre-mRNA splicing such as aberrant intron retention. Aberrant intron-retaining transcripts are exported to the cytoplasm, which may lead to mislocalization of their bound/cognate RBPs (e.g. SFPQ and FUS) from the nucleus to the cytoplasm in ALS. It follows that ASOs targeting aberrant splicing events might influence RBP nucleocytoplasmic distribution, but more experiments are required to demonstrate that these two phenomena are causally related. Figure created with BioRender.com
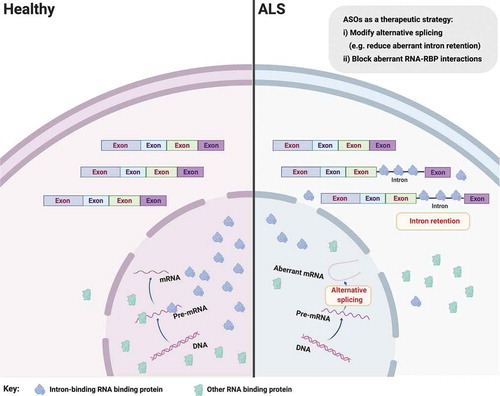
Figure 2. RNA binding protein mislocalization and aggregation in ALS. Defective compartmentalization of RBPs leads to their cytosolic accumulation and nuclear depletion in ALS, including TDP-43, SFPQ and FUS. RBPs can undergo liquid-liquid phase separation, however some RBPs abnormally form distinct conformations of amyloid cross β sheets and aggregates, inducing native monomers to misfold in ALS. Aggregation-prone RBPs can spread from cell to cell in a prion-like fashion. Therapeutic approaches perturbing protein mislocalization and misfolding, promoting the clearance of abnormally oligomerized proteins, or preventing the intercellular spread of these proteins are promising therapeutic strategies in ALS. Figure created with BioRender.com
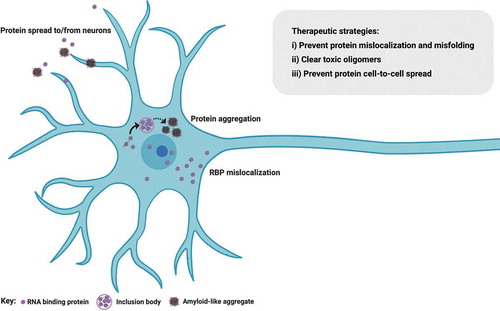
Figure 3. Glial involvement in ALS. Glia exhibit cell autonomous and non-cell autonomous neurodegeneration in ALS. As the disease progresses, astrocytes and microglia likely transition from an initial neuroprotective state to a toxic pro-inflammatory state in ALS with fewer neurotrophic factors and more neurotoxic factors secretion, such as inflammatory cytokines (TNF, IL-1, etc.), prostaglandin D2 PGD2 [Citation120,Citation127]. Reduced expression and activity of the astrocytic glutamate transporter EAAT2 influences motor neuron excitability. Oligodendrocytes fail to support axons of motor neurons through the disruption of lactate transport and normal myelination. Upon aging, glia-specific genes, but not neuron-specific genes, shift strikingly their regional expression patterns. A viable therapeutic approach might be to invoke the neuroprotective capacity of astrocytes or microglia. Similarly, cellular (glial) transplants may be a tractable therapy in ALS by promoting survival of juxtaposed motor neurons. Figure created with BioRender.com
![Figure 3. Glial involvement in ALS. Glia exhibit cell autonomous and non-cell autonomous neurodegeneration in ALS. As the disease progresses, astrocytes and microglia likely transition from an initial neuroprotective state to a toxic pro-inflammatory state in ALS with fewer neurotrophic factors and more neurotoxic factors secretion, such as inflammatory cytokines (TNF, IL-1, etc.), prostaglandin D2 PGD2 [Citation120,Citation127]. Reduced expression and activity of the astrocytic glutamate transporter EAAT2 influences motor neuron excitability. Oligodendrocytes fail to support axons of motor neurons through the disruption of lactate transport and normal myelination. Upon aging, glia-specific genes, but not neuron-specific genes, shift strikingly their regional expression patterns. A viable therapeutic approach might be to invoke the neuroprotective capacity of astrocytes or microglia. Similarly, cellular (glial) transplants may be a tractable therapy in ALS by promoting survival of juxtaposed motor neurons. Figure created with BioRender.com](/cms/asset/3f979417-a02a-433e-8289-d99cda654fe4/iett_a_1805734_f0003_oc.jpg)
Figure 4. Integration of models for ALS therapeutic study. An integrated approach for the discovery of high confidence therapeutic candidates for ALS. Patient iPSC-derived region-specific neurons and glia for primary discovery with mouse transgenic models and human postmortem tissue for secondary validation to identify promising candidates for further mechanistic and/or therapeutic evaluation. Insights gained from in vivo studies are then used to improve or modify in vitro model systems to better portray the disease and/or focus on different targets. Figure created with BioRender.com