
Figures & data
Figure 1. Life cycle of high-risk Human Papillomavirus (hrHPV) in cervical epithelia and the progression from infection to cervical cancer. 1A) First, hrHPV enters through a microwound to the basal membrane of the cervical epithelia, where the infected cells are maintained as a reservoir. The expression of E1 and E2 proteins drives the initial viral amplification in the lower layers of the epithelia. When these cells divide, the expression of E6 and E7 proteins stimulates cell proliferation (orange cells with a light orange nucleus) to the mid-layers of the epithelia. In the mid-layers, the expression of E4 with the help of E1, E2, and E5 facilitates higher viral DNA amplification (orange cells with an enlarged and irregular nucleus, known as koilocytotic cells). Finally, the cells leave the cell cycle (E4+) and express L1 and L2 proteins, allowing the packaging of the amplified virus (orange cells with a dark orange nucleus) and the subsequent release. Gene expression of viral proteins (right arrow), viral DNA amplification, and cell proliferation (left arrows) start from low levels (light orange) at the basal layers and go to high levels (dark orange) at the mid or upper layers of the cervical epithelia in hrHPV-infected women. 1B) After an hrHPV infection, the virus induces cell proliferation, and the infected tissue can progress to CIN. The first lesions, low-grade intraepithelial lesions (LSIL, CIN1), might regress after one year. However, they can also progress to high-grade intraepithelial lesions (HSIL, CIN2 – CIN3) and then to cervical cancer. CIN: cervical intraepithelial neoplasia
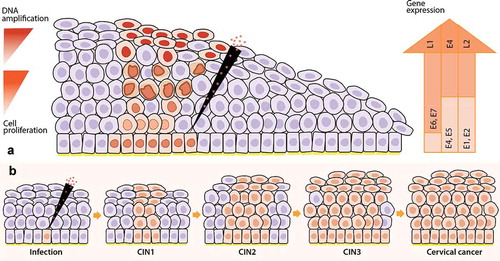
Figure 2. Viral biomarkers during HPV infection and progression to cervical cancer. After HPV infection at the basal membrane in the epithelia (virus detectable on DNA level), the virus will replicate its genome at low copy numbers, maintaining a latent infection in the basal keratinocytes. Next, the infection will switch to a productive infection where the virus expresses its genome, while keratinocytes differentiate, forming koilocytotic cells (E4+) and finally resulting in the release of the virus in the upper layers of the epithelia, enabling viral load assessment. After viral release, virions can re-infect the basal membrane leading to a persistent infection. Persistent infections present a higher risk of undergoing HPV genome integration to the host genome in the epithelia, resulting in cervical intraepithelial neoplasia (CIN) and the progression of these lesions cause cervical cancer. Nevertheless, the lesions can also regress thanks to the response of the host immune system that generates infection clearance. Approximately, in 70% of the cases, hrHPV16 and 18 genotypes are the causative agents of the progression to cervical cancer, which makes viral genotyping an essential biomarker. Besides, the progression of HPV infection to cervical cancer is highly related to the viral oncoproteins E4 and E6/E7; therefore, the analysis of their protein and transcriptional levels, respectively, is important as a biomarker testing for the disease. This infection progression is also associated with the HPV DNA methylation patterns that are higher in later stages of the lesions and cervical cancer
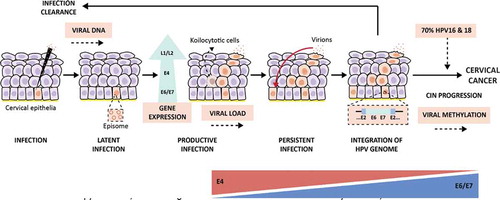
Figure 3. Downstream cellular effects of hrHPV E6/7 upregulation. Once expressed, viral E7 interferes with the pRb pathway by binding it and, hence, provoking the release of the E2F transcription factor. E2F will translocate to the nucleus where it will transcribe different proliferation-promoting genes; among these: MCM2 and TOP2A are overexpressed. The E7-pRb binding will avoid the negative feedback loop essential for the p16INK4A regulating protein (p16), which will be overexpressed to compensate for the following high proliferation level. At the same time, E6 binds p53 by promoting its degradation, which will cause the release of the Sp1 transcription factor, and also the impairment of p53-mediated interference with the pRb pathway. In fact, p53 prevents the phosphorylation of pRb; thus, the consequent activation of E2F. This event leads to the inhibition of apoptosis and transcription of Sp1-associated genes, like DNA methyltransferase enzymes (DNMTs) that will enhance the methylation of various promoters associated with HPV-mediated processes. The combination of these changes will provoke the boosted proliferation of the cell, which will be a signal for the production of Ki-67, a nuclear protein. In addition, E6 will upregulate the expression of TERT and TERC, essential for repairing telomeres. Soon, the cell could gain the chromosomal regions containing these two elements (3q and 5p), leading to genomic instability. It is believed that these events will facilitate viral genome integration, resulting in transforming cells. The abnormally over-expressed molecules involved in this process represent useful cellular biomarkers. TERC: telomerase RNA component; TERT: telomerase reverse transcriptase gene; MCM2: minichromosome maintenance protein 2; TOP2A: topoisomerase II alpha; CDK4/6: cyclin-dependent kinase 4 and 6
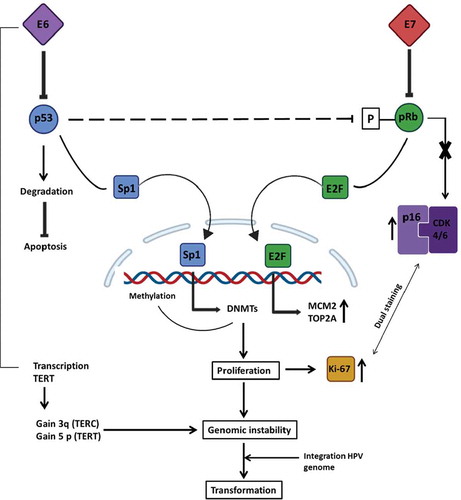
Figure 4. Mechanism of regulation mediated by p16INK4A during healthy conditions and HPV infection. In healthy non-HPV-infected cells (left), p16INK4A (p16) serves as a fundamental regulator of the cell cycle induction. When a cell has to go through G1/S transition, the cyclin-dependent kinases 4 and 6 (CDK4/6) forms a complex with cyclin D. The entire complex phosphorylates the retinoblastoma protein (pRb), which, in normal conditions, binds the transcription factor E2F. The phosphorylation mediated by the CDK4/6-cyclinD complex provokes E2F release, which promotes the transcription of those essential genes for the G1/S transition phase. At the same time, when the same healthy cell has to go towards cell cycle arrest, the regulatory protein p16 forms a complex with CDK4/6, preventing its binding to cyclin D and, hence, the transcription of E2F-correlated genes. However, in an HPV-infected cell (right), the viral protein E7 competes for the binding of pRb, and their linkage permits the release of E2F and the transcription of G1/S-genes. The expression of p16 will try to regulate the entire process, but the transcription factor release does not depend on CDK4/6-cyclinD phosphorylation; instead, it depends on E7 viral activity. Negative feedback mediated by pRb-E2F controls the expression of p16; hence, since this complex no longer exists, the loop is disrupted, and this will result in overexpression of p16 even if its regulatory activity is not accomplished
Figure 5. Viral and cellular biomarkers for detecting specific stages of cervical intraepithelial lesions after a hrHPV infection. After hrHPV infection, the cervical epithelia infected starts to proliferate, finally getting transformed to cervical intraepithelial neoplasia (CIN) 1, 2, and 3. Then from CIN3, it progresses to cervical cancer. According to our review, several viral and cellular biomarkers may be useful for detecting different stages of CIN. hrHPV-DNA primary test is the best test so far to apply in population-based screening. Then in CIN1, besides the hrHPV DNA testing we can also use genotyping; for CIN2: TOP2A/MCM2, p16/Ki-67 staining, 3q gain in the chromosome, Viral load, hrHPV transcriptional status (E6/E7 mRNA), and E4 protein, with the latter being able to stratify the heterogenous CIN2 group in safe lesions (E4 positive) and clinically significant lesions (E4 negative); and for CIN3: Host DNA methylation(m), miRNAs, hrHPV genotyping, and Viral DNA methylation (m)
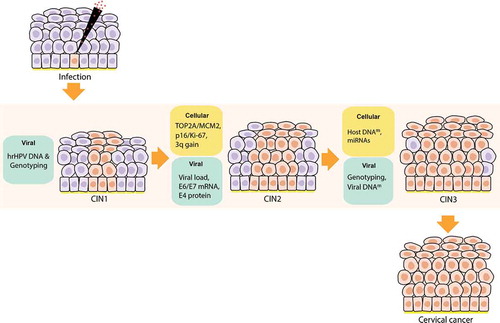
Table 1. Existing and candidate biomarkers for cervical cancer screening and triage