Figures & data
Figure 1. PI3K/AKT/mTOR signalling pathway. Growth factors bind to tyrosine kinase receptors (RTK) or G protein-coupled receptors (GPCR), activating PI3K heterodimers of the IA and IB families, respectively. Activated PI3K phosphorylates PIP2 into PIP3, while PTEN dephosphorylates PIP3. The activation of PIP3 attracts PDK1 and AKT to the plasma membrane. AKT can be phosphorylated by PDK1 at the T308 site, while mTORC2 phosphorylates the S473 site. Afterwards, the activated AKT can activate multiple downstream targets. The phosphorylation of tuberous sclerosis 2 (TSC2) by AKT can inhibit the TSC1/TSC2 complex, leading to indirect activation of mTORC1 by blocking the negative regulation of the Ras homolog (Rheb) by TSC1/2. S6 kinase 1 protein (S6K1) and eucaryotic initiation factor 4E-binding protein (4E-BP1) are downstream targets of mTORC1, which can control protein synthesis. In addition, AKT can also phosphorylate IKK, MDM2, FoxO1 to regulate cell survival, and GSK3β to regulate glucose metabolism.
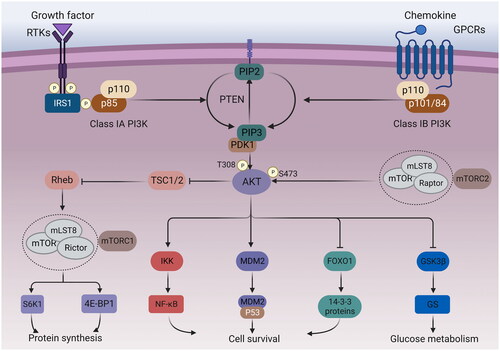
Table 1. A summary of major PI3K inhibitors.
Figure 2. Structures of pan-PI3K inhibitors.
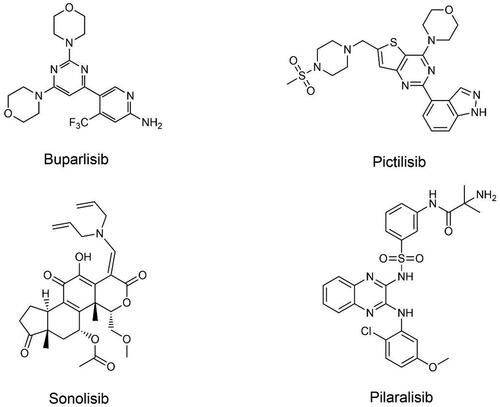
Figure 3. Structures of PI3K isoform-specific inhibitors.
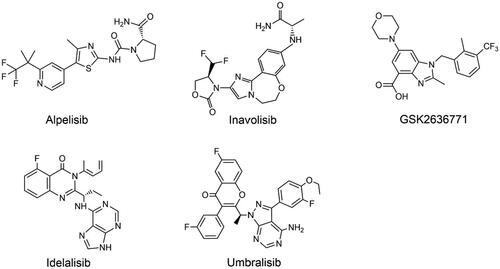
Figure 4. Structures of dual PI3K/mTOR or dual PI3K inhibitors.
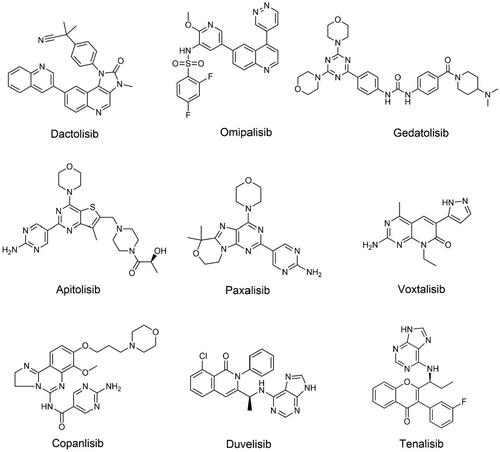
Figure 5. PIKK mediated DNA damage response (DDR) inactivation regulates the immune response. PIKK members ATM, ATR, and DNA-PK have the ability to promote cell cycle arrest and DNA repair. DNA-PK, via the Ku heterodimer, binds to double-strand broken DNA ends, promoting non-homologous end-joining (NHEJ). The MRN complex recruits ATM to single-strand broken DNA, which is then repaired via homologous recombination (HR). In response to HR, ATR is recruited to single-stranded DNA wrapped by replicating protein A (RPA) for DNA repair. DNA damage response (DDR) gene inactivation leads to the accumulation of chromosomal fragments in the cytoplasm, which activates the cyclic GMP-AMP synthase-interferon gene stimulator (cGAS-STING) pathway. STING activates interferon regulatory factor 3 (IRF3) transcription by binding to and activating tank-binding kinase 1 (TBK1). The type I IFN response is upregulated by the chemokine C-X-C motif, chemokine ligand 10 (CXCL10) and chemokine receptor 5 (CCL5), thereby promoting the infiltration of effector T cells into tumours. In addition, the expression of programmed cell death 1 ligand 1 (PD-L1) was upregulated. Secreted IFN-α and IFN-β promoted the antigen-presenting ability of dendritic cells.
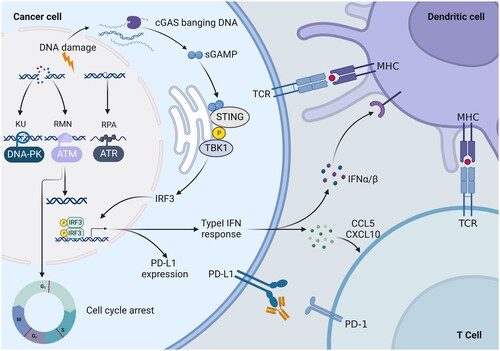
Figure 6. Structures of PIKK family members ATR, ATM, and DNA-PKcs inhibitors.
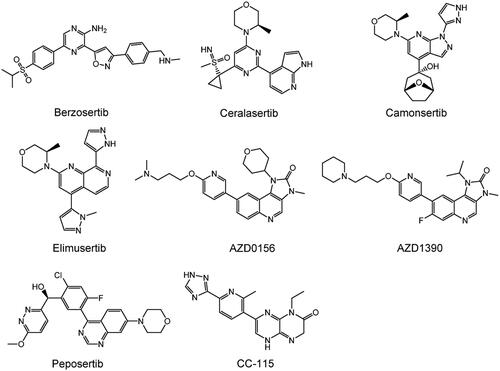
Table 2. A summary of major PIKK inhibitors.
Figure 7. Structures of PIKK family members mTOR inhibitors.
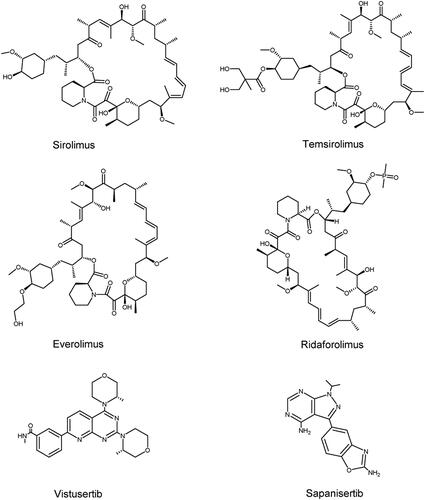
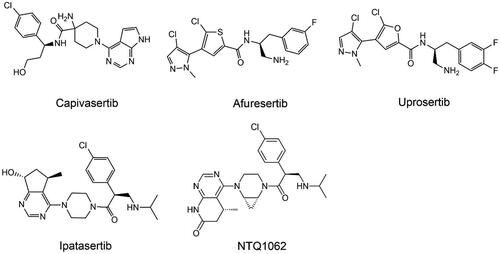
Table 3. A summary of major AKT inhibitors.
Figure 8. Structures of AKT allosteric Inhibitors.
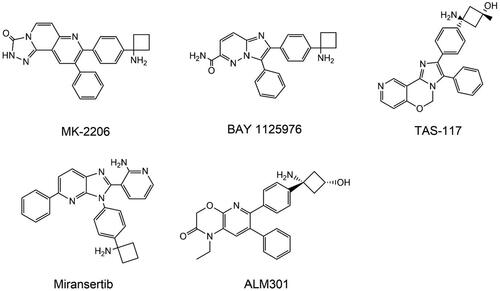
Figure 9. Structures of AKT competitive inhibitors.