Figures & data
Figure 1. Approach to identification of small molecule inhibitors of IRES-mediated translation. (A) The 5′-untranslated region of the human IGF1R mRNA. (B) Reporter constructs used to genetically engineer T47D human breast carcinoma cells for use in the high throughput screen to identify compounds that selectively interfere with function of the IGF1R IRES. The bicistronic construct used for the screen contains the full-length IGF1R 5′-untranslated sequence (1,040 nucleotides including the IRES, GenBank: NG_009492.1), positioned between the Renilla and firefly luciferase coding sequences. Cells stably transfected with the reporter construct containing the IGF1R 5′-UTR (in monocistronic context) from which the core functional IRES has been deleted were used for the counterscreen. (C) Scatter plot of representative raw data from one of the pilot HTS assays that preceded the full high-throughput screen / counterscreen. Relative inhibition of IRES activity (firefly bioluminescence signal generated from cells stably transfected with the bicistronic IRES reporter construct) is plotted vs. relative inhibition of signal generated by the cells stably transfected with the monocistronic control (no IRES) construct. (D) Summary of compound progression path for identification of small molecule IRES inhibitors. (E). Structure of IRES inhibitor lead compound P (cpd_P) = N-(4-anilinophenyl)-N′-[2-(4-chlorophenyl)ethyl]thiourea, MW 381; and candidate lead (false positive) cpd_T = N-[5-(5-isopropyl-1,3-benzoxazol-2-yl)-2-methoxyphenyl]butanamide, MW 352. Two closely related analogs of cpd_P are also utilized, in which the chlorine substituent is replaced by either a methoxy group (P-2) or a fluorine atom (P-3).
![Figure 1. Approach to identification of small molecule inhibitors of IRES-mediated translation. (A) The 5′-untranslated region of the human IGF1R mRNA. (B) Reporter constructs used to genetically engineer T47D human breast carcinoma cells for use in the high throughput screen to identify compounds that selectively interfere with function of the IGF1R IRES. The bicistronic construct used for the screen contains the full-length IGF1R 5′-untranslated sequence (1,040 nucleotides including the IRES, GenBank: NG_009492.1), positioned between the Renilla and firefly luciferase coding sequences. Cells stably transfected with the reporter construct containing the IGF1R 5′-UTR (in monocistronic context) from which the core functional IRES has been deleted were used for the counterscreen. (C) Scatter plot of representative raw data from one of the pilot HTS assays that preceded the full high-throughput screen / counterscreen. Relative inhibition of IRES activity (firefly bioluminescence signal generated from cells stably transfected with the bicistronic IRES reporter construct) is plotted vs. relative inhibition of signal generated by the cells stably transfected with the monocistronic control (no IRES) construct. (D) Summary of compound progression path for identification of small molecule IRES inhibitors. (E). Structure of IRES inhibitor lead compound P (cpd_P) = N-(4-anilinophenyl)-N′-[2-(4-chlorophenyl)ethyl]thiourea, MW 381; and candidate lead (false positive) cpd_T = N-[5-(5-isopropyl-1,3-benzoxazol-2-yl)-2-methoxyphenyl]butanamide, MW 352. Two closely related analogs of cpd_P are also utilized, in which the chlorine substituent is replaced by either a methoxy group (P-2) or a fluorine atom (P-3).](/cms/asset/99709bae-baa0-4d1d-906c-583e53232fca/kcbt_a_1071729_f0001_b.gif)
Figure 2. Reporter analyses demonstrate selective inhibition of IRES-mediated translation by candidate lead compounds and analogs. T47D breast tumor cells stably transfected with the bicistronic reporter construct containing the IGF1R 5′-UTR / IRES or the monocistronic control construct (no IRES) were seeded (in parallel) in 24-well plates and allowed 48 h to recover and resume proliferation, then treated with increasing concentrations (0 - 20 μg/ml) of IRES inhibitor lead cpd_ P (A, B), analogs P-2 (C, D) or P-3 (E, F), or candidate lead cpd_T (G, H) as indicated, under either Low (0.5%) serum (A,C,E,G) or Full (10%) serum (B, D, F, H) conditions. Following 24 h incubation, cells were harvested, lysates prepared, and firefly and Renilla luciferase activities assayed. Three parameters are assessed: firefly (2nd cistron) translation, as a direct readout of IRES-mediated translation; Renilla (1st cistron) translation, mediated by cap-dependent ribosomal scanning; and M, representing translation of the independent monocistronic control (no IRES). The dotted horizontal line is indicative of the firefly luciferase activity measured following treatment with cycloheximide (100 μg/ml) for 24 h. All data ± standard error.
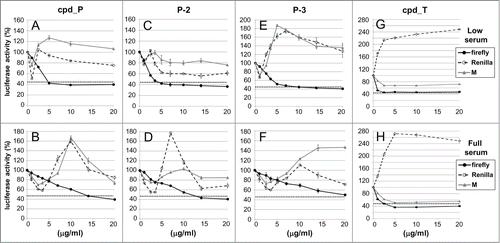
Figure 3. Activity of IRES inhibitors against the endogenous IGF1R IRES. (A) T47D cells were seeded in 6-well plates and allowed 48 h to recover prior to treatment with increasing concentrations of IRES inhibitor cpd_P or vehicle control as indicated. Following 72 h continuous exposure to the compound, cells were harvested, whole cell lysates prepared, equivalent aliquots separated by SDS/PAGE, and analyzed by western blot for IGF1R. (B) Titration of IRES inhibitor cpd_P on regeneration of IGF1R following trypsin catabolism. T47D human breast tumor cells were trypsinized and reseeded (in full serum) into 6-well plates and exposed immediately to increasing concentrations of IRES inhibitor cpd_P. Following 24 h incubation, cells were harvested, whole cell lysates prepared, equivalent aliquots separated by SDS/PAGE, and assayed by western blot for IGF1R. Robust regeneration of trypsin- catabolized IGF1R is observed within 24 h in vehicle (DMSO) treated cells, however, this is completely blocked in the presence of cpd_P at ≥ 5 μg/ml. Trypsin: lysate prepared from cells immediately following trypsinization. The arrow marks the position of intact membrane-bound IGF1R (β subunit). *asterisk marks the position of trypsin-catabolized IGF1R. Results confirm activity of cpd_P against the endogenous IGF1R IRES in native (genetically-unmodified) tumor cells. (C) The trypsin catabolism / regeneration assay was used to titrate IRES inhibition by analog P-3 and candidate lead cpd_T.
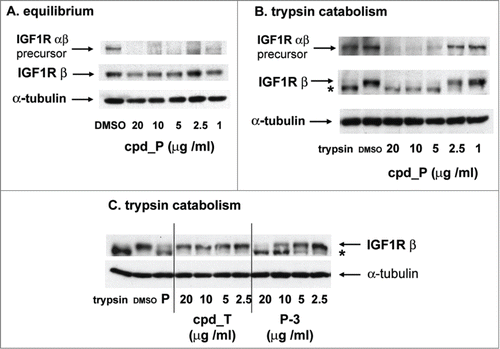
Figure 4. IRES inhibitor cpd_P blocks de novo synthesis of IGF1R in response to acute serum deprivation. (A) SUM159 breast tumor cells were seeded in 6-well plates and allowed 48 h in full serum media to recover and resume proliferation. Then, the standard growth media (which includes 5% fetal calf serum and 5 μg/ml insulin) was replaced with media containing only 0.5% FCS and no supplemental insulin, along with IRES inhibitor lead cpd_P, analog P-2 or P-3, candidate lead cpd_T (each at 10 μg/ml), or vehicle (0.1% DMSO) control. After 24 h, cells were harvested, whole cell lysates prepared, equivalent aliquots separated by SDS/PAGE and analyzed by western blot for IGF1R. (B) Trypsin catabolism combined with serum deprivation. SUM159 breast tumor cells were trypsinized and reseeded into 6-well plates and incubated immediately in the presence of compounds as indicated, in low serum media (0.5% FCS, no supplemental insulin). Robust regeneration and upregulation of IGF1R is observed within 24 h in vehicle (DMSO) treated cells, however, this is completely blocked in the presence of IRES inhibitor cpd_P or analogs P-2 or P-3. *asterisk marks the position of trypsin-degraded IGF1R.
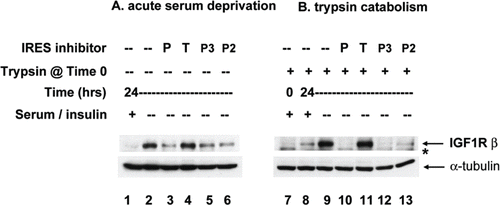
Figure 5. IRES inhibitor cpd_P is highly active against Myc and is distinguished from other translationally-active drugs. (A) SUM159 breast tumor cells were subjected to acute serum / insulin deprivation (0.5% FCS, no supplemental insulin) and simultaneously treated X 24 h with IRES inhibitor cpd_P (10 μg / ml), rapamycin (100 nM in lanes 3, 6; 200 nM in lane 4), cycloheximide (100 μg / ml), anisomycin (10 μM), or combinations of these reagents as indicated. Whole cell lysates were prepared, equivalent aliquots separated by SDS/PAGE, and analyzed by western blot for IGF1R and c-Myc. Insulin receptor, driven by its own unique IRES,Citation71,72 and mrtl, a Myc-related protein which does not require use of an IRES,Citation73 serve as controls. IRES inhibitor cpd_P differentially modulates translation of the 2 Myc isoforms, decreasing abundance of p64 (oncogenic), while increasing synthesis of p67 (growth-inhibitory). In contrast, rapamycin secondarily stimulates synthesis of both isoforms of Myc, while cycloheximide and anisomycin completely eliminate all Myc protein. (B) Western blot results obtained for Myc after 24, 48, or 72 h treatment of SUM159 breast tumor cells with variable concentrations (1.0 - 10 μg/ml) of cpd_P in full serum.
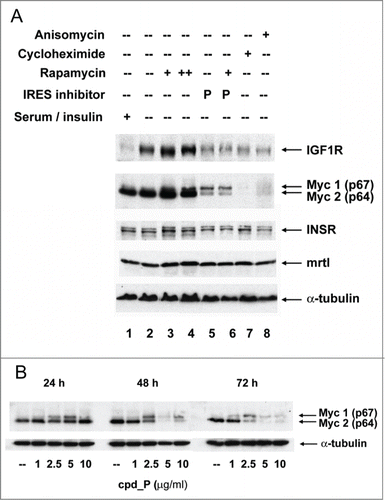
Figure 6. Phenotypic consequences of IRES inhibition. (A) Viability of SUM159 breast tumor cells assessed following treatment with increasing concentrations of cpd_P for periods of 24 to 120 h. Cell survival is presented relative to the cell number at initiation of treatment (time 0 = 100%) ± standard error. (B) The viability time course titrations of IRES inhibitor cpd_P in SUM159 breast tumor cells were repeated under Low serum (0.5%) conditions (all data ± standard error). (C) Clonogenic survival assay. SUM159 breast tumor cells were seeded at low density, allowed 48 h to recover, then treated with increasing concentrations of cpd_P in full serum for 72 h. Media was then changed, compound removed, and cells allowed an additional 120 h to recover and form colonies. Cultures were stained with MTT to enhance visualization of colonies.
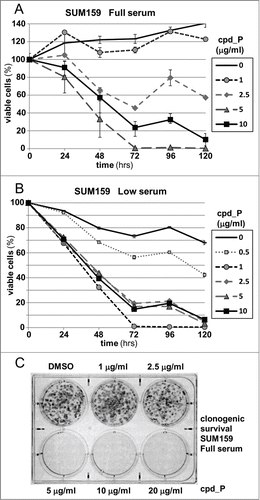
Figure 7. RNase protection analysis of c-myc mRNA in human breast tumor cells treated with IRES inhibitor cpd_P. (A) Diagram of the human c-myc locus and the series of antisense probes designed to assess the abundance of each of the 4 c-myc mRNA isoforms. Sequence coordinates are as described in Gazin et alCitation74 (GenBank: X00364). (B) RNase protection time course assay. SUM159 breast tumor cells were treated with IRES inhibitor cpd_P (10 μg/ml in full serum) and harvested at selected time points. 5 μg of total RNA recovered from each sample (or tRNA as negative control) was hybridized with each of the probes overnight at 42°C. An extensive series of pilot experiments had indicated that P0 is the predominant c-myc mRNA isoform present in these cells, and that good resolution could be obtained by combining the P1/P2 and P3/exon 2 probes while analyzing the P0 probe separately. Following hybridization, samples were digested with RNase A / T1, recovered by ethanol precipitation, and separated on 5% acrylamide, 8M urea denaturing gels. Only 1/20th of the amount of probe included in each digested sample was loaded in the positive control (undigested) lanes. The P0 transcript is expected to protect a fragment of 149 nucleotides. Protected fragments of 504 and 342 nt are expected for the P1 and P2 transcripts. P3-initiated transcripts would be contiguous with exon 2, and should protect the full-length intron 1 – exon 2 probe. With exon 2 beginning at 4506, the spliced c-myc mRNAs (cumulatively) are expected to protect a fragment of 249 nt. The asterisk marks the position of a faint band which maps to the 3′ end of exon 1 and apparently represents a rare splicing event for the P0 transcript (additional data not shown). The two gels were run in parallel and exposed to autoradiography for the same amount of time. Pr = full-length probe (undigested). t = tRNA (negative control).
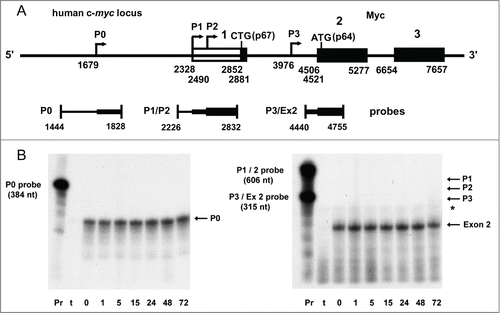
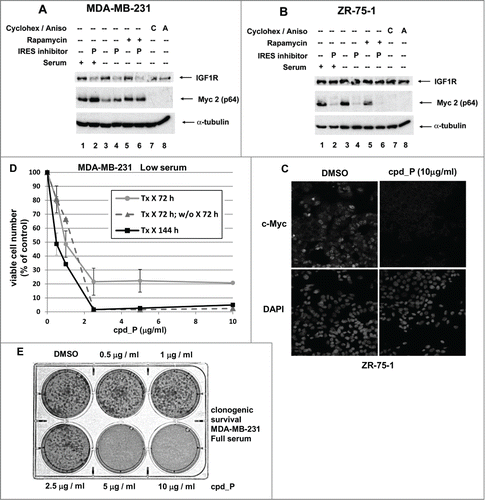
Figure 8. Myc translation-regulatory status evaluated on the basis of sensitivity to IRES inhibition. (A) MDA-MB-231 or (B) ZR-75-1 breast tumor cells were treated for 24 h with IRES inhibitor cpd_P (10 μg/ml), rapamycin (100 nM), cycloheximide (100 μg / ml), anisomycin (10 μM), or combinations of these reagents as indicated, in either Full serum (10% for MDA-MB-231, 20% for ZR-75-1) or Low serum (0.5%) conditions. Cells were harvested, whole cell lysates prepared, equivalent aliquots separated by SDS/PAGE, and analyzed by western blot for IGF1R and c-Myc. (C) ZR-75-1 breast tumor cells were seeded in 8-well chamber slides, treated with IRES inhibitor cpd_P (10 μg/ml) or vehicle control (DMSO 0.1%) for 24 h in low (0.5%) serum, then stained for Myc, following PFA (2% × 15 min) fixation and low (0.2% × 10 min) Triton X-100 permeabilization. Confocal imaging demonstrates dramatic loss of Myc from cpd_P-treated cells. DAPI staining in the associated panels marks the locations of all nuclei in each field. (D) MDA-MB-231 breast tumor cells were treated with cpd_P (0 - 10 μg/ml in low serum) for 72 or 144 h, or treated for 72 h followed by 72 h incubation in absence of compound (washout). The graph displays viability outcomes for the cpd_P-treated cells relative to DMSO (vehicle)-treated controls (100%) ± standard error. (E) Clonogenic survival assay. MDA-MB-231 cells were seeded at low density, allowed 48 h to recover, then treated with increasing concentrations of cpd_P in full (10%) serum for 96 h. Media was then changed, compound removed, and cells allowed an additional 96 h to recover and form colonies. Cultures were stained with MTT to enhance visualization of colonies.