Figures & data
Figure 1. PARP1 regulated HMGB1 polyADP-ribosylation, which subsequently promoted HMGB1 acetylation. (a) Silencing FIP200 reduced chemotherapy-induced HMGB1 acetylation. Jurkat cells were transfected with control or FIP200 shRNA and then treated with or without DNR (0.4 µM) for 24 hours. The cell lysates were pulled down with an HMGB1 antibody and immunoblotted with anti-acetylated lysine and HMGB1 antibodies. β-actin was used as a loading control. (b) PARP1 was required for HMGB1 PARylation and subsequently promoted the acetylation of HMGB1 in vitro. HMGB1 was PARylated and successively acetylated as described in the Materials and Methods. The reactions were analyzed by western blot. Quantified data are presented (PARylation-HMGB1 or Acetylation-HMGB1/HMGB1, n > 3, *p < 0.05). HI, heat-inactivation. (c) Chemotherapy-induced autophagy was accompanied by polyADP-ribosylation and acetylation modification of HMGB1. Jurkat cells and RS4:11 cells were treated with or without DNR (0.4 μM) for 24 hours. The cell lysates were subjected to immunoprecipitation with an HMGB1 antibody followed by western blot analysis, and then the acetylation or PARylation level was measured using specific antibodies against acetylated lysine or PARylated. β-actin was used as a loading control. Quantified data are presented (PARylation-HMGB1 or Acetylation-HMGB1/HMGB1/β-actin, n > 3, *p < 0.05).
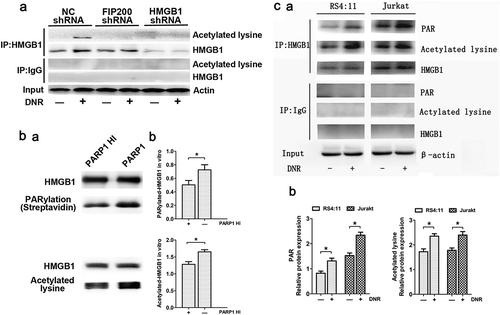
Figure 2. SIRT6 served as an upstream signal for the activation of PARP1 through monoADP-ribosylation. (a) PARP1 or SIRT6 in Jurkat cells was successfully depleted. Jurkat cells were transfected with control, PARP1 or SIRT6 shRNA. The mRNA and protein levels of PARP1 or SIRT6 were assessed via RT-qPCR or western blot analyses, respectively (n > 3, *p < 0.05). AU, arbitrary unit. The mock group was set as 1. (b) The activation of PARP1 induced by DNR was depressed in SIRT6-silencing cells. Cells were exposed to 0.4 μM DNR. The activity of PARP1 was measured by cell ELISA (n > 3, *p < 0.05 compared with the control group without DNR treatment). (c) SIRT6 and PARP1 formed a protein complex, and SIRT6 catalyzed the monoADP-ribosylation of PARP1. Jurkat cells were treated with or without DNR (0.4 μM) for 24 hours. The cell lysates were immunoprecipitated with an anti-SIRT6 antibody and immunoblotted with an anti-PARP1 antibody.
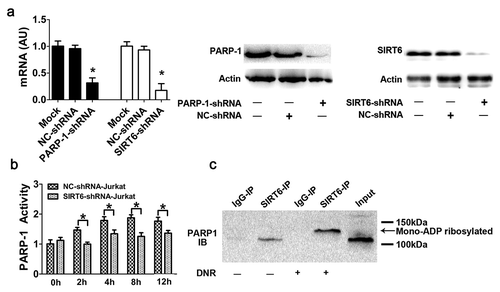
Figure 3. Silencing of SIRT6 or PARP1 inhibited the translocation of HMGB1 and suppressed chemotherapy-induced autophagy in leukemic cells. (a) The chemotherapy-induced autophagy and transition of HMGB1 were depressed in SIRT6 or PARP1 shRNA-treated cells. Jurkat cells were transfected with control, PARP1 or SIRT6 shRNA followed by the presence or the absence of DNR (0.4 μM) or CQ (10μM) for 24 hours. The cell lysates were subjected to western blot to detect the expression of LC3-II/I and p62. Furthermore, the cell lysates were separated into cytosolic and nuclear fractions. Cytosolic and nuclear proteins were subjected to western blot and detected with an HMGB1 antibody. β-actin and lamin B were used as loading controls. Quantified data are presented (p62 or LC3-II/I/β-actin, nuc-HMGB1/lamin B, cyt-HMGB1/β-actin, n > 3, *p < 0.05). CQ: chloroquine, Cyt: cytoplasm, Nuc: nucleus. (b) Autophagosomes and autophagolysosomes in SIRT6 or PARP1 shRNA-treated cells were less numerous than in the NC control group during chemotherapy. Jurkat cells were transfected with control, PARP1 or SIRT6 shRNA followed by the presence or the absence of DNR (0.4 μM) for 24 hours. The cells were subjected to transmission electron microscopy to observe autophagosome-like structures (indicated by the red arrows). (c) The absence of SIRT6 or PARP1 depressed the translocation of HMGB1 from the nucleus to the cytoplasm. Jurkat cells were transfected with control, PARP1 or SIRT6 shRNA followed by the presence or the absence of DNR (0.4 μM) for 24 hours. Intracellular HMGB1 was stained by immunofluorescence and subjected to confocal microscopic analysis to detect the location of HMGB1 (green: HMGB1; blue: nucleus). Quantified data are presented. UT: untreated group.
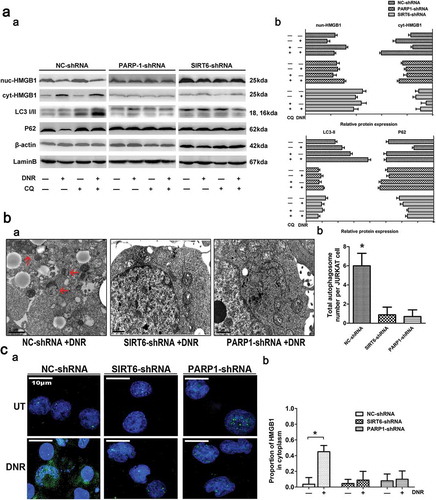
Figure 4. The SIRT6-PARP1 complex was related to the polyADP-ribosylation and acetylation of HMGB1. Jurkat cells were transfected with control, PARP1 or SIRT6 shRNA with or without DNR (0.4 μM) treatment for 24 hours. The cell lysates were pulled down with an HMGB1 antibody and immunoblotted with anti-acetylated lysine, anti-PARylation or HMGB1 antibodies. β-actin was used as a loading control.
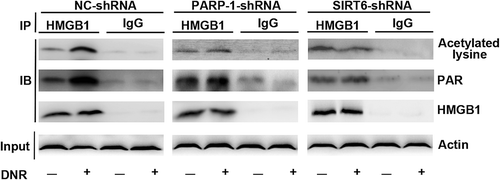
Figure 5. PARylation of HMGB1 facilitated its acetylation and promoted HMGB1 translocation-related autophagy. (a) HMGB1MT1, HMGB1MT2, HMGB1WT were successfully constructed. HMGB1MT1, HMGB1MT2, HMGB1WT, HMGB1NC and Jurkat cells were subjected to western blot analysis to detect the expression of lentivirus with a specific anti-Flag antibody. Tubulin was used as a loading control. (b) Jurkat cells were transfected with lentivirus, and then HMGB1NC, HMGB1MT1, HMGB1MT2 and HMGB1WT were treated with or without DNR (0.4 μM) for 24 hours. The cell lysates were pulled down with an HMGB1 antibody and immunoblotted with anti-acetylated lysine, anti-PARylation and HMGB1 antibodies. β-actin was used as a loading control. Quantified data are presented (PARylation-HMGB1 or Acetylation-HMGB1/HMGB1/β-actin, n > 3, *p < 0.05). (c) Jurkat cells were transfected with lentivirus, and then HMGB1NC, HMGB1MT1, HMGB1MT2 and HMGB1WT were treated with or without DNR (0.4 μM) for 24 hours. The cell lysates were subjected to western blot to detect the expression of LC3-II/I and p62. Furthermore, the cell lysates were separated into cytosolic and nuclear fractions. Cytoplasmic proteins were subjected to western blot and detected with an HMGB1 antibody. β-actin and lamin B were used as loading controls. Quantified data are presented.
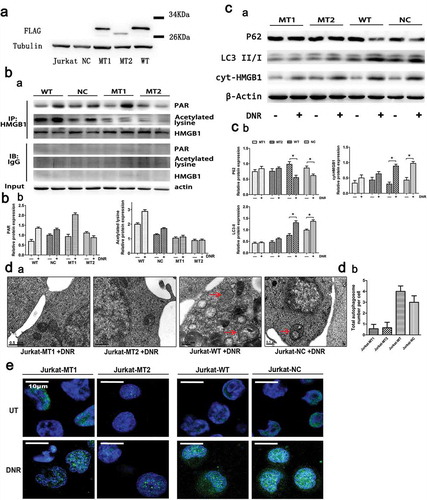
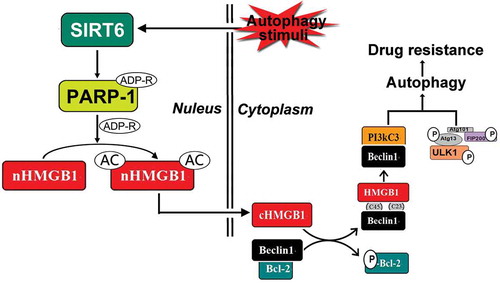
Figure 6. Schematic representation of the potential HMGB1-translocation related chemotherapy-induced autophagy in leukemia. Under stress condition, SIRT6 was activated, SIRT6 not only served as an upstream signal of PARP1, but also formed the complex with PARP1. SIRT6 activated PARP1 through monoADP-ribosylation and the activated PARP1 modified HMGB1 by polyADP-ribosylation, which subsequently enhanced the acetylation of HMGB1 and promoted the translocation of the HMGB1 from nucleus to cytoplasm. The translocation of HMGB1 was the most critical step during this process and mediated chemotherapy-induced autophagy in leukemic cells.