
Figures & data
Figure 1. Multiple genetic lesions impact the metabolism in cancer cells. Sustained activation of RTK pathways, when occurring concurrently with gain of Myc and loss of p53 functions, is responsible of metabolic challenges that allow cancer cells to proliferate under normoxia or hypoxia via increased glucose consumption, nucleotide-protein-lipid synthesis and glutamine anapleurosis. In addition, these pathways may confer resistance to oxidative stress.
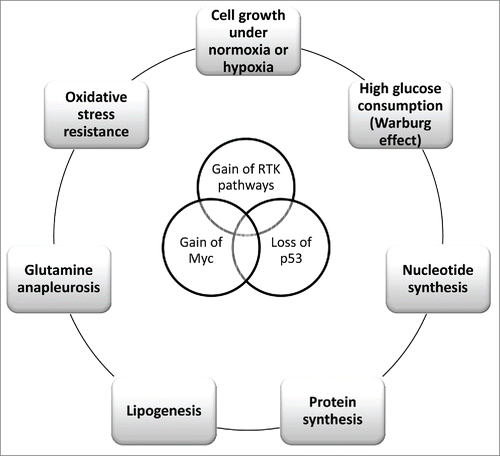
Figure 2. The genomic landscape in fusion-positive and -negative RMS. Fusion-positive RMS have a relatively low burden of somatic mutations; they are dominated by the fused Pax3-Foxo1 signature and high copy number of N-Myc gene. Instead, fusion-negative RMS are characterized by a number of different alterations, including mutations in the p53 pathway or cell cycle genes and gain of chromosomes. Both the fusion-positive and -negative subsets share the aberrant activation of RTK/RAS/PI3K axis as well as the IGF-2 overexpression due to LOI and LOH at 11p15.5 locus, respectively.
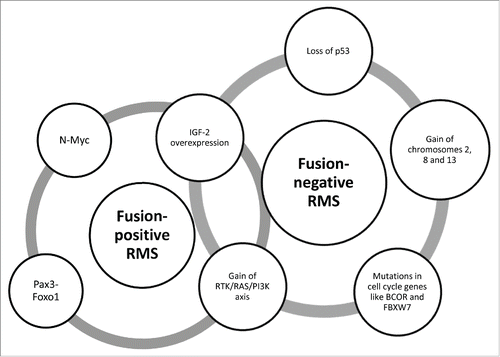
Figure 3. Molecular signatures influencing the metabolism in cancer and RMS cells. The cartoon depicts the molecular effectors that in cancer cells increase the glucose uptake and glycolytic consumption, nucleotide synthesis, lipogenesis, protein synthesis and glutamine anapleurosis. As observed in different tumors, RMS subsets share many of these molecular hallmarks; moreover, the more aggressive fusion-positive RMS express the Pax3-Foxo1 oncoprotein, which is actively involved in the transcription of different targets, including metabolic genes such as GLUT4 and CPT1A. In addition, Pax3-Foxo1 increases the expression of N-Myc and decreases that of PTEN. Thin arrows indicate the interconversion occurring between organic substrates, whereas thick arrows indicate the activating (in green) or inhibitory (in red) actions of the protein effectors on different processes through enzymes (blue box outlined) or transporters (red box outlined). Abbreviations: ACL, ATP citrate lyase; CPT1A, carnitine palmitoyl transferase 1A; FAS, fatty acid synthase; GLUTs, GLUT transporters; G6PD, glucose-6-phosphate dehydrogenase; HIF, hypoxia inducible factor; HK, glycolytic hexokinase; LDH, lactate dehydrogenase; MAPK, mitogen activated protein kinase; PDC, pyruvate dehydrogenase complex; PDK1, pyruvate dehydrogenase kinase isozyme 1; PFK, phosphofructokinase; PI3K, phosphatidylinositol-3-kinases; mTOR, mammalian target of rapamycin; RTK, tyrosine kinase receptors; PTEN, phosphate and tensin homolog deleted on chromosome 10; SREBPs, Sterol Regulatory Element-Binding Proteins.
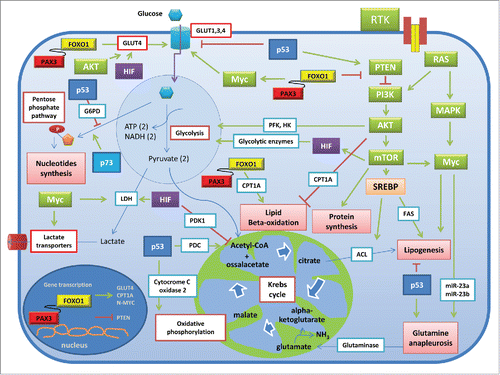
Table 1. List of the more recently completed phase I/II clinical trial studies on drugs against RMS.