Figures & data
Figure 1. Plk1 interacts with nuclear SREBP1 during mitosis. (A) HEK293 cells were transfected with FLAG-tagged nuclear SREBP1a (nSREBP1a), either wild-type (WT, amino acids 1–490) or the ΔC mutant (amino acids 1–417), lacking the entire C-terminal domain of nSREBP1a. Cell lysates were inmunoprecipitated with anti-FLAG antibodies. The amounts of immunoprecipitated Plk1 and nSREBP1a, and the levels of nSREBP1a and Plk1 in whole-cell lysates (WCL) were determined by Western blotting, with α-tubulin serving as a loading control. (B) HEK293 cells were transfected with FLAG-nSREBP1a, either WT or the S439A mutant. Cell lysates were immunoprecipitated with anti-FLAG antibodies. The amounts of immunocrecipitated Plk1 and nSREBP1a, and the levels of nSREBP1a, Plk1 and α-tubulin in cell lysates were determined by Western blotting. (C) Cell lysates from HeLa cells were used in peptide pull-down assays, using 2 peptides corresponding to residues 436–442 of human SREBP1a, either non-phosphorylated (Ref) or the same peptide phosphorylated on S439 (P-pept) (upper panel). Myc-PBD, either WT or a binding-deficient mutant (MT), was expressed in HEK293 cells, and the cell lysates were used in peptide pull-down assays, using the same 2 peptides as those described above (lower panel). The bound proteins were subjected to SDS/PAGE and Western blotting using 20% of input as control. (D) Recombinant nSREBP1a, either WT or the S439A mutant, was used in in vitro kinase assays in the absence or presence of recombinant Cdk1/cyclin B. The phosphorylated proteins were mixed with lysates of HEK293 cells expressing GFP-Plk1. The His-tagged nSREBP1a proteins were captured on NiTA-agarose, washed and resolved by SDS/PAGE, followed by Western blotting. The phosphorylation of S439 in nSREBP1a was monitored with a phosphorylation-specific antibody (pS439). (E) HeLa cells were synchronized at the G1/S transition by a double-thymidine protocol. Cells were collected after the second thymidine block (G1/S) or 14 h after the release into either nocodazole-containing media (Mit) or normal media (G1). Cell lysates were incubated with recombinant GST-PBD, either WT or binding-deficient mutant (MT), in the presence of glutathione beads. The amount of nSREBP1 associated with the glutathione beads was determined by Western blotting (left panel). The GST-PBD proteins were detected by coomassie brilliant blue staining (CBB). The levels of nSREBP1 and α-tubulin in cell lysates were determined by Western blotting (right panel) (F) HeLa cells were synchronized at the G1/S transition by a double-thymidine protocol. Cells were collected after the second thymidine block (G1/S) or 14 h after the release into either nocodazole-containing media (Mit) or normal media (G1). Cell lysates were immunoprecipitated with either a control antibody (Gal4, lanes 1–3) or SREBP1 antibody (lanes 4–6). The amounts of immunoprecipitated Plk1 and nSREBP1 were determined by Western blotting (left panel). The levels of nSREBP1, Plk1 and α-tubulin in cell lysates were determined by Western blotting (right panel).
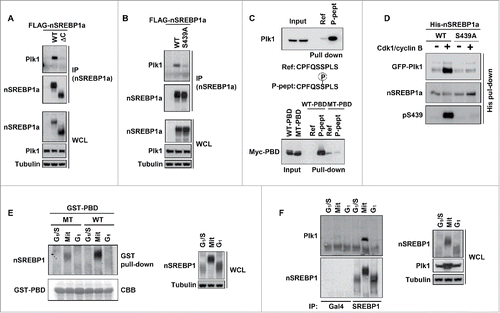
Figure 2. Plk1 phosphorylates T424, S467 and S486 in nuclear SREBP1 during mitosis. (A) In vitro kinase assay with recombinant nSREBP1a and Plk1. The levels and phosphorylation (pT424, pS467 and pS486) of nSREBP1a were monitored by Western blotting. (B) Recombinant nSREBP1a was used in in vitro kinase assays with mitotic HeLa extracts (Mit) in the absence or presence of the Plk1 inhibitor BTO-1 (5, 10 and 25 μM). The levels and phosphorylation (pT424, pS467 and pS486) of nSREBP1a were monitored by Western blotting. (C) Recombinant nSREBP1a was used in in vitro kinase assays with extracts from HeLa cells transfected with either control or Plk1 siRNA. The levels and phosphorylation (pT424, pS467 and pS486) of nSREBP1a were monitored by Western blotting (left panel). The efficiency of the Plk1 knockdown was monitored by Western blotting, with α-tubulin serving as a loading control (right panel). (D) Recombinant nSREBP1a, either WT or the S439A mutant, was used in in vitro kinase assays with mitotic HeLa extracts. The levels and phosphorylation (pT424, pS467 and pS486) of nSREBP1a were monitored by Western blotting. (E) HEK293 cells were transfected with nSREBP1a or nSREBP1c and left untreated or treated with nocodazole. After immunoprecipitation, the levels and phosphorylation (pT424, pS467 and pS486) of the respective nSREBP1 protein were determined by Western blotting. (F) HeLa cells were synchronized at the G1/S transition by a double-thymidine protocol. Cells were collected after the second thymidine block (G1/S) or 14 h after the release into either nocodazole-containing media (Mit) or normal media (G1). The levels and phosphorylation (pT424, pS467 and pS486) of nSREBP1 were determined by Western blotting following immunoprecipitation of SREBP1.
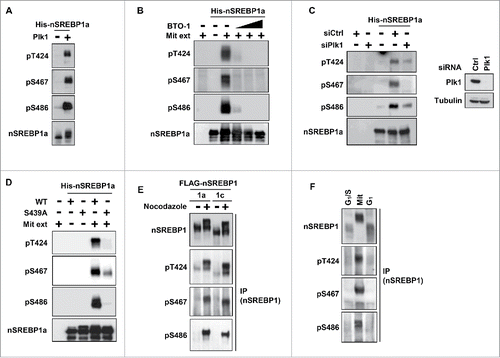
Figure 3. Plk1 phosphorylates SREBP1 in vivo. (A) HCT116 cells were synchronized at the G1/S transition by a double-thymidine protocol. After the first thymidine block, cells were transfected with control or Plk1 siRNA. Cells were collected at the indicated time points after release from the second thymidine block in media containing nocodazole. Cell lysates were analyzed by Western blotting with antibodies against the indicated proteins. The levels and phosphorylation (pT424, pS467 and pS486) of nSREBP1 were determined by Western blotting following immunoprecipitation of SREBP1. (B) HeLa cells were left untreated or treated with nocodazol to induce G2/M arrest. Where indicated, BI-2536 (250 nM) was added for the last 2 hours. The levels and phosphorylation (pT424, pS467 and pS486) of nSREBP1 were determined by Western blotting following immunoprecipitation of SREBP1. (C) HEK293 cells were transfected with nSREBP1a (left panel) or nSREBP1c (right panel) in the absence or presence of increasing amounts of WT or kinase-deficient (KD) Plk1. The levels of nSREBP1a, nSREBP1c, Plk1 and α-tubulin (loading control) were determined by Western blotting. (D) HEK293 cells were transfected with nSREBP1a in the presence of GFP (left panel) or GFP-Plk1 (right panel). Forty-eight hour after transfection, cells were treated with cycloheximide (CHX, 100 μgr/ml) for the indicated times. The levels of nSREBP1a, Plk1 and α-tubulin (loading control) were determined by Western blot analysis. The relative amount of nSREBP1a at each time point is plotted as percentage of the amount at the start of the assay (lower panel).
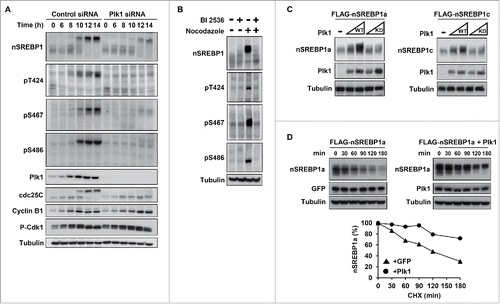
Figure 4. Plk1 regulates the stability and transcriptional activity of nuclear SREBP1. (A) HEK293 cells were transfected with nSREBP1a, either WT or the indicated mutants. The levels of nSREBP1a were determined by Western blotting, with α-tubulin serving as a loading control. (B) HEK293 cells were transfected with nSREBP1a, either WT or the 3A mutant (left panel) or the 3D and 3E mutants (right panel), in the presence of control or Plk1 siRNA. The levels of nSREBP1a, Plk1 and α-tubulin were monitored by Western blotting. (C) HepG2 cells were transfected with the indicated promoter reporter construct. Twenty-four hours after transfection, cells were treated with nocodazole for 16h and luciferase activity was measured. Where indicated, cells were treated with BTO-1 (50 μM) for the last 4h. The data represent the averages ± SD of three independent experiments. Statistical analyses were performed using a 2-tailed Student's t-test. For all tests, P-values lower than 0.05 were considered statistically significant. *P < 0 .05, **P < 0 .01 and ***P < 0 .001. (D) HepG2 cells were transfected with SYNSRE-luc and either control or Plk1 siRNA. Twenty-four hours after transfection, cells were treated with nocodazole and luciferase activity was measured. The data represent the averages ± SD of three independent experiments. Statistical analyses were performed using a 2-tailed Student's t-test. For all tests, P-values lower than 0.05 were considered statistically significant. *P < 0 .05, **P < 0 .01 and ***P < 0 .001. (E) HeLa cells were synchronized at the G1/S transition by a double-thymidine protocol. Cells were collected after the second thymidine block (G1/S) or 14 h after the release into nocodazole-containing media (Mit). Where indicated, cells were treated with BTO-1 (50 μM) for the last 4 h. Total RNA was used to determine the expression of the indicated genes by semi quantitative RT-PCR analysis.
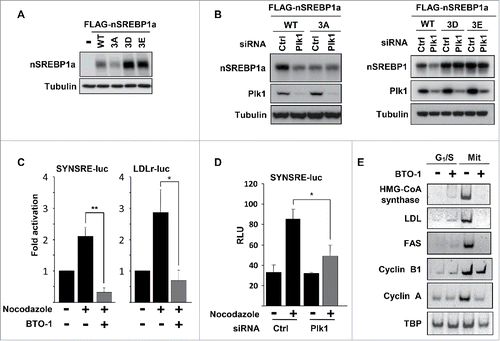
Figure 5. Plk1-mediated phosphorylation of nuclear SREBP1 prevents its degradation by Fbw7. (A) WT (Fbw+/+) or Fbw7 knockout (Fbw7−/−) HCT116 cells were treated in the absence or presence of nocodazole. The levels of SREBP1, Cyclin E, Plk1 and α-tubulin (loading control) were determined by Western blotting. The membrane-associated precursor (P) and nuclear (N) forms of SREBP1 are indicated. (B) HeLa cells were left untreated or treated with nocodazole to induce mitotic arrest. Where indicated, cells were treated with the proteasome inhibitor MG-132 (25 μM) for the last 4 h. The levels of nSREBP1 and α-tubulin were determined by Western blotting. (C) HEK293 cells were transfected with nSREBP1a in the absence or presence of Fbw7α and the cells were left untreated or treated with nocodazole. The levels of nSREBP1a, Fbw7α and α-tubulin were determined by Western blotting. (D) HEK293 cells were transfected with nSREBP1a, either WT or the 3A mutant, in the absence or presence of increasing amounts of Fbw7α. The levels of nSREBP1a, Fbw7α and α-tubulin were determined by Western blotting (upper panel). The percentage of nSREBP1a remaining is plotted against the concentration of Fbw7α (lower panel). The data represent the averages ± SD of three independent experiments. (E) HEK293 cells were transfected with nSREBP1a, either WT or the 3A mutant, in the absence or presence of Plk1 and Fbw7α. The levels of nSREBP1a, Plk1, Fbw7α and α-tubulin were monitored by Western blotting. (F) HEK293 cells were transfected with nSREBP1c, either WT or the 3D mutant, together with control or Fbw7 siRNA. The levels of nSREBP1c and α-tubulin were determined by Western blotting. (G) HEK293 cells were transfected with nSREBP1a and either left untreated or treated with nocodazole. Lysates from the transfected cells were mixed with recombinant GST-Fbw7α. Following capture of GST-Fbw7α and extensive washing, the bound proteins were subject to SDS/PAGE and the amount of nSREBP1a and Fbw7α were determined by Western blotting (upper panel). The amount of nSREBP1a and α-tubulin in whole cell lysates (WCL) were determined by Western blotting (lower panel). (H) HEK293 cells were transfected with FLAG-nSREBP1a, either WT or the T424D mutant. Cell lysates were immunoprecipitated with anti-FLAG antibodies. The amounts of immunoprecipitated Fbw7 and nSREBP1a (upper panel), and the levels of nSREBP1a, Fbw7 and α-tubulin in cell lysates (Input, lower panel) were determined by Western blotting. The band indicated by the asterisk corresponds to the IgG chain. (I) HeLa cell lysates were used in peptide pull-down assays, using 2 peptides corresponding to residues 422–442 of human SREBP1a, either phosphorylated on T426 and S430 (2xP-pept) or the same peptide phosphorylated on T424, T426 and S430 (3xP-pept). The bound proteins were subjected to SDS/PAGE and Western blotting using 20% of input as control.
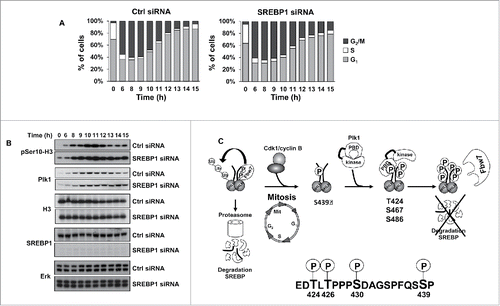
Figure 6. SREBP1 inactivation results in a delay in mitotic exit. (A) HCT116 cells were synchronized at the G1/S transition by a double-thymidine protocol. After the first thymidine block, cells were transfected with control (Ctrl) or SREBP1 siRNA. Cells were collected at the indicated time points after release from the second thymidine block and used for FACS analysis. The cell cycle distribution at each time point corresponds to the average of 3 independent experiments. (B) HCT116 cells were treated as in (A) and collected at the indicated time points after the second thymidine block. Cell lysates were analyzed by Western blotting with antibodies to the indicated proteins. (C) Phosphorylation-dependent stabilization of nSREBP1 during mitosis. Cdk1 phosphorylates the C-terminus of nSREBP1 thereby creating a docking site for Plk1. Plk1 binds nSREBP1 during mitosis, resulting in the phosphorylation of at least 3 residues in the C-terminus of nSREBP1. Phosphorylation of these residues attenuates the interaction between nSREBP1 and Fbw7, thereby blocking Fbw7-dependent degradation of nSREBP1 during mitosis.